Capítulo 5
CAPÍTULO 5
Pesquisa del cáncer colorrectal en grupos de riesgo aumentado
VIGILANCIA COLONOSCÓPICA POSTPOLIPECTOMÍA
El adenoma es el principal precursor del cáncer colorrectal (CCR) y la lesión colónica más frecuentemente hallada en las colonoscopias realizadas por pesquisa.1
En este capítulo se abordará acerca de la necesidad de un plan de vigilancia con colonoscopia, luego de la polipectomia y de un correcto diagnóstico histológico.
Luego de la extracción de adenomas del colon, de un tercio a la mitad de los pacientes van a presentar adenomas en colonoscopias posteriores realizadas a los tres años. Más aún, entre un 0,3 y 0,9% de ellos presentarán cáncer dentro de los 5 años.2-9 Muchas de estas lesiones representan pólipos pasados por alto en colonoscopias basales (pólipos perdidos), lo que nos remarca la importancia de realizar una colonoscopia de alta calidad.9-12
El objetivo principal de la vigilancia colonoscópica es prevenir el desarrollo de CCR a través de la resección de nuevos adenomas o de aquellos pasados por alto en estudio previos. La colonoscopia no es perfecta y no todos los CCR son prevenidos por este método, por lo cual la vigilancia también está dirigida a detectar CCR en estadios tempranos y, de este modo, mejorar la sobrevida de estos pacientes.7,13
La colonoscopia es un procedimiento invasivo con un riesgo de complicaciones bajo, aunque no despreciable, perforación en 0,06% de estudios diagnósticos y 2% en estudios con polipectomía y hemorragia (0,2%-2,7%, dependiendo del tamaño de la lesión).14-17 También representa un alto costo para los sistemas de salud. En EE.UU., el 22% de las colonoscopias realizadas en mayores de 55 años corresponden a estudios de vigilancia y en muchas ocasiones se encuentra sobre indicada.9,18 Por todo lo mencionado, es fundamental dirigir la vigilancia a aquellos pacientes que realmente se beneficien de ella, y con la mínima frecuencia necesaria para proveer protección contra el desarrollo de CCR con una baja tasa de complicaciones. A continuación describiremos los distintos factores de riesgo para el desarrollo de CCR, que determinarán posteriormente la necesidad de vigilancia.
FACTORES DE RIESGO PARA EL DESARROLLO DE LESIONES NEOPLÁSICAS AVANZADAS (LNA) Y CCR LUEGO DE LA RESECCIÓN BASAL DE ADENOMA
Factores relacionados al procedimiento endoscópico
- Calidad de la colonoscopia
La sensibilidad de la colonoscopia no alcanza al 100% ni siquiera en colonoscopias completas.
Tanto adenomas como LNA y cáncer pueden ser pasados por alto, sobre todo en estudios con preparaciones deficientes.9,12,19,20 La tasa de pólipos perdidos en colonoscopias tándem (dos estudios realizados en el mismo día) varía entre un 25 a 50%. Más preocupante aún es el hecho de que el 6% de las lesiones >1cm y un 4% de CCR también son pasados por alto por la colonoscopia.13
Estos hallazgos son similares a la tasa de detección de adenomas y de lesiones avanzadas en los primeros estudios de vigilancia postpolipectomía, lo que sugiere que la mayoría de las lesiones encontradas en los estudios de vigilancia temprana corresponden a pólipos perdidos.9,11,21 La estratificación del riesgo para realizar vigilancia colonoscópica surge de asumir que los pacientes con múltiples adenomas o lesiones avanzadas presentan mayor riesgo de desarrollar nuevas lesiones de similares características, sumado a un mayor riesgo de otras lesiones pasadas por alto durante la colonoscopia. Por este motivo resulta esencial lograr llevar a cabo colonoscopias de alta calidad, con preparaciones adecuadas, un examen exhaustivo de la mucosa y resecciones completas de las lesiones encontradas.11,20
Las colonoscopias de vigilancia también deben ser realizadas con los máximos estándares de calidad.9,11,12,22 La mayor parte de los cánceres de intervalo en pacientes, realizando colonoscopias de vigilancia, surge de lesiones resecadas en forma incompleta o pasadas por alto durante el estudio.7,23-25
Las colonoscopias de alta calidad realizadas con poca frecuencia son más efectivas en prevenir
el CCR que aquellas de baja calidad realizadas frecuentemente.
Las colonoscopias se deben llevar a cabo con preparaciones satisfactorias, en forma completa y con un adecuado tiempo de retirada (al menos 6 minutos), que permita una correcta evaluación de la mucosa.
A aquellos pacientes con estudios en los que no se logra la intubación cecal, se les recomienda repetir el estudio o realizar algún estudio alternativo complementario, especialmente en pacientes con alto riesgo de CCR.9,22
La decisión dependerá también de las características del paciente (edad, grupo de riesgo), de los hallazgos del estudio, del riesgo de repetir el estudio, del estado de salud y de la preocupación del paciente. También, de factores locales tales como las listas de espera y la posibilidad de ser realizado por otro endoscopista de mayor experiencia.
B. Adenomas resecados en forma incompleta
Un cuarto de todos los cánceres colorrectales que se diagnostican antes de los tres primeros años de realizada una colonoscopia se desarrollan en el sitio de la resección previa de una lesión.9,11,12,23
Las lesiones planas grandes resecadas mediante fragmentos, como mencionamos previamente, presentan un mayor riesgo de tejido neoplásico residual. Por este motivo es importante la re examinación del sitio de polipectomía a los 3 meses. Las áreas pequeñas de tejido residual pueden resecarse endoscópicamente. En caso de presencia de áreas extensas de tejido residual se recomienda la resección quirúrgica.
Factores relacionados con las características de los adenomas
A. Número de adenomas
La presencia de múltiples adenomas en la colonoscopia de base es el predictor más consistente para el hallazgo de lesiones avanzadas y cáncer en colonoscopias de vigilancia.
Un meta-análisis de estudios de vigilancia con colonoscopia demostró que los pacientes con más de 3 adenomas en el estudio basal duplican el riesgo de encontrar LNA.26 Un pool análisis posterior de Martínez y colaboradores27 que incluyó 9.167 pacientes confirmó la asociación de más de 3 pólipos con el hallazgo de lesiones avanzadas y cáncer; sin embargo, el riesgo era 4 veces mayor ante la presencia de 5 o más adenomas.
La alta tasa de detección de lesiones avanzadas y cáncer en colonoscopias de vigilancia luego de la resección de múltiples adenomas probablemente resulte de: un alta tasa de pólipos perdidos sumado al potencial de dichos adenomas de desarrollar lesiones avanzadas.
B. Tamaño del adenoma
Un meta-análisis publicado recientemente mostró que aquellos pacientes con al menos un pólipo mayor a 1cm y a 2cm en colonoscopias de base tenían 2 y 4 veces más de chance, respectivamente, de presentar lesiones avanzadas y cáncer en estudios de vigilancia.27
Se recomienda en lo posible la medición del pólipo en mm y con algún instrumento de medición,
ya que la visión endoscópica suele ser muy subjetiva. En lo posible se deberá tomar en cuenta la medición del patólogo. Este aspecto es importante ya que va a definir el intervalo de vigilancia.
C. Histología
El componente histológico velloso o túbulo-velloso en adenomas presentes en las colonoscopias basales es un predictor inconsistente de posteriores lesiones avanzadas y cáncer. Tomando en cuenta ciertas características que de por sí representan un factor de riesgo contundente –como el número y el tamaño de los pólipos–, sumado al pobre acuerdo interobservador de las clasificaciones histológicas para definir adenoma velloso, la histología individualmente no debería tomarse como un factor de riesgo significativo para el posterior desarrollo de las lesiones mencionadas.
D. Grado de displasia
Algunos estudios observacionales sugirieron una relación significativa entre displasia de alto grado (DAG) en las lesiones basales y el hallazgo de LNA y cáncer en estudios posteriores.26 Sin embargo, un pool análisis que incluyó 6 estudios, luego de ajustar por diferentes factores de riesgo –tales como tamaño y número de pólipos–, la DAG no demostró ser un factor de riesgo independiente.27 Debemos analizar esta evidencia con precaución ya que la prevalencia de DAG en pólipos menores a 10mm es sólo del 1% y, por lo tanto, los estudios publicados carecen del poder estadístico necesario para confirmar o descartar esta asociación. La vigilancia luego de la extracción de pólipos pequeños con DAG deberá ser discutida localmente por cada grupo dependiendo de su situación y las posibilidades.
Factores relacionados con las características de los pacientes
A. Edad y sexo
Aunque la edad avanzada ha demostrado ser un factor de riesgo para LNA, ésta no se considera un motivo para intensificar la vigilancia colonoscópica.28 La colonoscopia resulta menos efectiva y más riesgosa en pacientes ancianos. Además, el tiempo de progresión de un adenoma a cáncer, que es de aproximadamente 10 a 20 años, es similar a la expectativa de vida de un paciente de 75 años. Esto sugiere que la mayoría de los pacientes a esta edad no se va a beneficiar de realizar vigilancia con colonoscopia.
Si bien el sexo masculino ha demostrado ser un factor de riesgo moderado en algunos estudios, aún no es claro si este factor debería influir en la vigilancia.28
B. Historia familiar
Varios estudios demostraron que existe una mayor prevalencia de adenomas en colonoscopias
basales de pesquisa en aquellos pacientes con antecedentes familiares de CCR.29,30 Si bien algunos estudios pequeños reportaron un mayor riesgo de LNA en la vigilancia colonoscópica de aquellos pacientes con antecedentes familiares de CCR, un estudio prospectivo de 1.287 pacientes y un meta-análisis posterior no pudieron confirmar esta asociación.27,31 En consecuencia, no existe evidencia consistente para sustentar la recomendación de intensificar la vigilancia en pacientes con antecedentes familiares de CCR, a menos que se sospeche una condición hereditaria.
Recomendaciones e intervalos de vigilancia para los pólipos colorrectales
Las recomendaciones de varios países Europeos, EE.UU. y de Asia han definido tres grupos de riesgo (bajo, intermedio y alto) basados en el número y en las características de los adenomas detectados en la colonoscopia de base.9,11,32-36 La correcta estratificación, según el riesgo de los pacientes y la adecuación de los intervalos de vigilancia, reduciría la realización de estudios innecesarios, los costos y, finalmente, las complicaciones. Las recomendaciones para la vigilancia postpolipectomía sólo se aplican después de una colonoscopia de base de alta calidad con la eliminación completa de todas las lesiones neoplásicas detectadas.9,11,12,36
A. Grupo de bajo riesgo
Varios estudios realizados en pacientes bajo vigilancia colonoscópica identificaron un grupo de pacientes con un bajo riesgo de desarrollar LNA y CCR.11,18,31,37-39 Excepto en uno de estos estudios hubo acuerdo en que la presencia de 1 o 2 adenomas confiere un riesgo bajo de LNA posteriores.31,40 Como mencionamos previamente, hubo desacuerdo en cuanto al tamaño y a la histología, que son variables estrechamente relacionadas entre sí y resulta difícil separar el efecto de cada variable.
Un estudio realizado en EE.UU. comparó el riesgo de LNA en vigilancia colonoscópica entre pacientes con 1-2 adenomas y aquellos sin ninguna lesión en colonoscopia de base.18 Aunque los resultados demostraron que luego de 5 años no hubo diferencia entre ambos grupos, confiriendo un bajo riesgo a los pacientes con 1-2 adenomas, debemos aclarar que en este estudio existió una importante pérdida de seguimiento de los pacientes incluidos.
Un reciente meta-análisis de Hassan y colaboradores40 muestra que los pacientes con adenomas de bajo riesgo en el estudio inicial tienen una mayor probabilidad de LNA metacrónica que en los pacientes sin adenomas al inicio, aunque el riesgo absoluto fue bajo en ambos grupos.
El estudio nacional del pólipos en EEUU demostró que el riesgo acumulativo de CCR 6 años después de la colonoscopia basal era un 75% más bajo que en la población general.41 En este estudio también se identificaron como pacientes de alto riesgo aquellos con más de 3 pólipos o aquellos con pólipos de gran tamaño, y como de bajo riesgo a aquellos con 1 a 2 adenomas pequeños.
Por lo mencionado se considera como de bajo riesgo para el desarrollo de LNA o CCR a aquellos
pacientes con 1 o 2 adenomas pequeños,independientemente de la histología en estudios basales.
El beneficio de la vigilancia con colonoscopia en este grupo de pacientes es pobre y probablemente
poco costo-efectivo. La recomendación para este grupo es la de realizar pesquisa de rutina: 5 a 10 años.
B. Grupo de riesgo intermedio
Ha sido demostrado que aquellos pacientes con 3 o más adenomas en estudios basales y particularmente si uno de ellos es mayor a 10mm presentan un mayor riesgo de LNA y CCR.27,39
En el estudio nacional de pólipos, el 9% de los pacientes con más de 3 adenomas y el 5% de aquellos con adenomas grandes en colonoscopias de base desarrollaban LNA en el primer estudio de vigilancia.41 En comparación con lo mencionado, sólo el 1% de los pacientes que presentaban un adenoma pequeño desarrollaban LNA.
Otro estudio de la Cleveland Clinic demostró que, en comparación con la presencia de 1 o 2 adenomas en estudios basales, la presencia de más de 4 adenomas incrementaba 5 veces el riesgo de desarrollar LNA, y la presencia de al menos un adenoma mayor a 10mm incrementaba 10 veces este riesgo.39
Dos estudios evaluaron el riesgo de desarrollar CCR a largo plazo luego de la extracción de pólipos colónicos grandes. En el primero de ellos, el riesgo de CCR fue 3 veces mayor en aquellos pacientes a los que se les resecó un adenoma >10 mm que en la población general.42 En el segundo estudio, el riesgo fue 4 veces mayor luego de la extracción de adenomas >10 mm o con componente velloso, y de hasta 7 veces mayor si los adenomas también eran múltiples.43
Ya que la presencia de más de 3 adenomas o de 1 adenoma >10 mm confiere un incremento del riesgo de desarrollar LNA o CCR, se considera
mandatorio realizar vigilancia colonoscópica en este grupo. Los resultados del estudio nacional de pólipos en EEUU sugieren que un intervalo de 3 años
desde la colonoscopia de base es adecuado para la mayor parte de los pacientes en este grupo.41
C. Grupo de alto riesgo
Estudios recientes mostraron que una proporción no despreciable de pacientes realizando estudios de vigilancia a los 3 años aún presentaban un mayor riesgo de desarrollar neoplasias.11 En el pool análisis de Martínez y colaboradores27 la presencia de 5 adenomas o uno >20 mm incrementaba este riesgo 4 veces. Los pólipos perdidos y las resecciones incompletas probablemente sean la explicación de este incremento de riesgo.
Por lo tanto, aquellos pacientes con más de 5 adenomas o uno mayor a 20mm se beneficiarían de realizar una vigilancia colonoscópica
temprana a los 12 meses. Ya que 12 meses es poco tiempo para el desarrollo de lesiones de novo, el objetivo primordial va a ser detectar y resecar
lesiones sincrónicas. Por este motivo la realización de una colonoscopia de alta calidad es fundamental.
Ajuste de la vigilancia
A. Significado de un estudio de vigilancia normal
Un estudio retrospectivo sobre 389 pacientes bajo vigilancia con colonoscopia anual mostró una tasa de detección de adenomas de sólo el 10% si la colonoscopia previa era normal, de 40% si tenía pólipos y de 70% si tenía múltiples pólipos.44 Otro estudio demostró que sólo una colonoscopia de vigilancia negativa no descarta el subsecuente hallazgo de pólipos en posteriores estudios de vigilancia.45
Por ello, y ante la existencia de evidencia de pobre calidad metodológica, debemos asumir que aquellos pacientes de riesgo alto e intermedio continúan teniendo un alto riesgo pese a la presencia de un colonoscopia de vigilancia negativa. Luego de una segunda colonoscopia negativa podemos tener más confianza de no haber pasado por alto adenomas y, por ende, de que el riesgo va a ser menor.11
Ante la falta de evidencia, en pacientes de riesgo intermedio se recomienda extender el intervalo a cinco años luego
de un primer estudio de vigilancia negativo. Luego de dos estudios consecutivos negativos, la vigilancia puede suspenderse.
En los pacientes de alto riesgo, luego de dos estudios anuales consecutivos negativos, se recomienda prolongar el intervalo a 2 años. Ante la falta de evidencia no se recomienda discontinuar la vigilancia.
B. Suspensión de la vigilancia
El beneficio de la realización de vigilancia debe ser medido según la edad y las comorbilidades del paciente. La edad que se considera como límite es la de 75 años; sin embargo, va a depender de la voluntad, de las comorbilidades y del tipo de pólipo.9
C. Desarrollo de síntomas en intervalos de vigilancia
Ya que la vigilancia reduce el riesgo de desarrollar neoplasias pero no lo elimina, la presencia de síntomas sugestivos nuevos debe ser tenida en cuenta para su estudio. 9
Vigilancia colonoscópica luego de la resección de otro tipo de lesiones
A. Resección de cáncer colorrectal pT1
Existen dos razones para realizar vigilancia luego de la extracción de este tipo de lesiones: evaluar la recurrencia y la detección de lesiones sincrónicas.
Por su naturaleza, los pólipos cáncer son lesiones con un alto riesgo de desarrollar lesiones neoplásicas. Por este motivo se recomienda realizar una estrategia de vigilancia similar a la de los pacientes de alto riesgo. Esta recomendación asume que el estudio basal fue de alta calidad, que la resección del pólipo cáncer fue completa y que el sitio de resección fue re examinado previamente.
La recomendación para la vigilancia luego de la resección quirúrgica con intención curativa de un cáncer de colon es realizar una colonoscopía de calidad perioperatoria (previo a la misma o de 3 a 6 meses luego si no se pudo realizar) con el objetivo de descartar lesiones sincrónicas o pólipos con potencial de malignidad. Y la vigilancia luego de la cirugía: al año, 4 años, 9 años posoperatorio y luego cada 5 años. Si se detectara alguna lesión en dichas endoscopías se adecuará la vigilancia en función de dichas lesiones.46


B. Adenomas serratos
Los adenomas serratos son lesiones que antes eran clasificados como pólipos hiperplásicos, hoy se los considera como lesiones premalignas (adenomas serratos sésiles y tradicionales) y que podrían ser precursores de hasta un 15% del CCR esporádico. Hoy se sabe que su vía se desarrollo hacia CCR es distinta que la clásica APC del adenoma → carcinoma. En éste se observa mutación del BRAF, metilación de MLH1 e inestabilidad microsatelital. Por su morfología plana, su dificultad diagnóstica y su localización en el colon derecho se los considera unos de los principales responsables de la falla de la colonoscopía en el prevención del cáncer de colon del lado derecho.11,12,47 La estrategia de vigilancia para pacientes con pólipos serrados propuesta por la mayoría del sociedades científicas internacionales se basan de estudios de pobre calidad metodológica.9,11,12
Estrategia de vigilancia con colonoscopía para adenomas aserrados:
- Bajo riesgo:
- 1-2 pólipos aserrados, adenoma serrato < 10 mm, sin displasia → pesquisa de rutina (5-10 años)
- Riesgo intermedio:
- 3-4 pólipos serratos < 10 mm sin displasia o
- 1-4 pólipos serratos < 20 mm sin displasia
- Pólipos serratos con displasia citológica → colonoscopía a los 3 años
- Alto riesgo:
- 5 o más pólipos serratos
- Pólipo serrato > 20 mm → colonoscopía al año
La presencia de adenomas de alto riesgo en forma sincrónica predice la presencia de lesiones avanzadas
metacrónicas porlo que la vigilancia debiera ser aún más estricta con menor tiempo de intervalo entre endoscopías.
BIBLIOGRAFÍA
- Imperiale, T. F.; Wagner, D. R.; Lin, C. Y.; Larkin, G. N.; Rogge, J. D. (2000). Risk of advanced proximal neoplasms in asymptomatic adults according to the distal colorectal findings, N. ENgl. J. Med., vol.343, Nº 3, pp. 169-174.
- Nozaki, R.; Takagi, K.; Takano, M.; Miyata, M. (1997). Clinical investigation of colorectal cancer detected by follow-up colonoscopy after endoscopic polypectomy, Dis Colon Rectum, vol. 40, Nº 10 Suppl, pp. S16-622.
- Alberts, D. S.; Martinez, M. E.; Hess, L. M. et al. (2005). Phase III trial of ursodeoxycholic acid to prevent colorectal adenoma recurrence, J. Natl. Cancer Inst., vol 97, Nº 11, pp. 846-853.
- Schatzkin, A.; Lanza, E.; Corle, D. et al. (2000). Lack of effect of a low-fat, high-fiber diet on the recurrence of colorectal adenomas. Polyp Prevention Trial Study Group, N.ENGL.J. Med, vol. 342, Nº 16, pp. 1149-1155.
- Lund, J. N.; Scholefield, J. H.; Grainge, M. J. et al. (2001). Risks, costs, and compliance limit colorectal adenoma surveillance: lessons from a randomized trial, Gut, vol. 49, Nº 1, pp. 91-96.
- Baron, J. A.; Cole, B. F.; Sandler, R. S. et al. (2003). A randomized trial of aspirin to prevent colorectal adenomas, N. ENgl.J.Med., vol. 348, Nº 10, pp. 891-899.
- Robertson, D. J.; Reenberg, E. R.; Beach, M. et al. (2005). Colorectal cancer in patients under close colonoscopic polypectomy, Dis Colon Rectum, vol. 36, Nº 12, pp. 1126-1131.
- Arber, N.; Eagle, C. J.; Spicak, J. et al. (2006) Celecoxib for the prevention of colorectal adenomatous polyps, N.Engl.J.Med, vol 355, Nº 9, pp. 885-895.
- Lieberman DA, Rex DK, Winawer SJ et al. Guidelines for Colonoscopy Surveillance after Screening and Polypectomy: A Consensus Update by the US Multi-Society Task Force on Colorectal Cancer Gastroenterology 2012; 143:844–857.
- Rex, D. K.; Bond, J. H.; Winawer, S. et al. (2002). Quality in the technical performance of colonoscopy and the continuous quality improvement process for colonoscopy: recommendations of the U.S Multi-Society Task Force on Colorectal Cancer, Am. J. Gastroenterol., vol. 97, Nº 6, pp. 1296-1308.
- Hassan C, Quintero E, Dumonceau J M et al. Post-polypectomy colonoscopy surveillance: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy 2013; 45: 842–851
- Bogie R, Sanduleanu S. Optimizing post-polypectomy surveillance: A practical guide for the endoscopist. Digestive Endoscopy 2016; 28: 348–359.
- Bressler, B.; Paszat, L. F.; Vinden, C.; Li, C.; He, J.; Rabeneck, L. (2004). Colonoscopic miss rates for right-sided colon cancer: a population-based analysis, Gastroenterology, vol. 127, Nº 2, pp. 452-456.
- Macrae, F. A.; Tan, K. G.; Williams, C. B. (1983). Towards safer colonoscopy: a report on the complications of 5000 diagnostic or therapeutics colonoscopies, Gut, col 24, Nº 5, pp. 376-383.
- Nivatvongs, S. (1986). Complications in colonoscopic polypectomy. An experience with 1,555 polypectomies, Dis. Colon Rectum, vol. 29, Nº 12, pp. 825-830.
- Waye, J. D.; Lewis, B. S.; Yessayan, S. (1992). Colonoscopy: a prospective report of complications, J. Clin. Gastroenterol, vol. 1, Nº 4, pp. 347-351.
- Rosen, L.; Bub, D. S.; Reed, J. F.; III & Nastasee, S. A. (1993). Hemorrahge following colonoscopic polypectomy, Dis.Colon Rectum, vol. 36, Nº12, pp. 1126-1131.
- Lieberman, D. A.; Weiss, D. G.; Harford, W. V. et al. (2007). Five year colon surveillance after screening colonoscopy, Gastroenterology, vol. 133, Nº 4, pp. 1077-1085.
- Rex, D. K. (2000). Colonoscopic withdrawal technique is associated with adenoma miss rates, Gastrointest. Endosc., vol. 51, Nº 1, pp. 33-36.
- Lebwohl B, Kastrinos F, Glick M, et al. (2011). The impact of suboptimal bowel preparation on adenoma miss rates and the factors associated with early repeat colonoscopy. Gastrointest Endosc.; 73(6):1207-1214.
- Pohl H, Robertson DJ. (2010) Colorectal cancers detected after colonoscopy frequently result from missed lesions. Clin Gastroenterol Hepatol; 8:858–864
- Short MW, Layton MC, Teer BN and Domagalski JE. Colorectal Cancer Screening and Surveillance. Am Fam Physician. 2015; 91(2):93-100.
- Pabby, A.; Schoen, R. E.; Weissfeld, J. L. et al. (2005). Analysis of colorectal cancer ocurrence during surveillance colonoscopy in the dietary Polyp Prevention Trial, Gastrointest. Endosc. vol. 61, Nº 3, pp. 385-391.
- Baxter NN, Sutradhar R, Forbes SS, Paszat LF, Saskin R, Rabeneck L. Analysis of administrative data finds endoscopista quality measures associated with postcolonoscopy colorectal cancer. Gastroenterology. 2011; 140(1):65-72.
- Corley DA, Jensen CD, Marks AR, et al. Adenoma detection rate and risk of colorectal cancer and death. N Engl J Med. 2014; 370 (14):1298-1306.
- Saini, S. D.; Kim, H. M.; Schoenfeld, P. (2006). Incidence of advanced adenomas at surveillance colonoscopy in patients with a personal history of colon adenomas: a meta-analysis and systematic review, Gastrointest Endosc. vol. 64, Nº 4, pp. 614-626.
- Martinez, M. E.; Baron, J. A.; Liebermen, D. A. et al. (2009). A pooled analysis of advanced colorectal neoplasia diagnoses after colonoscopic polypectomy, Gastroenterology, vol. 136, Nº 3, pp. 832-841.
- Yamaji, Y.; Mitsushina, T.; Ikuma, H.; Watabe, H.; Okamoto, M.; Kawabe, T.; Wada, R.; Doi, H.; Omata, M. (2004). Incidence and recurrence rates of colorectal adenomas estimated by annually repeated colonoscopies on asymptomatic Japanese, Gut, vol.53, Nº4, pp.568-572.
- Bonelli, L.; Martines, H.; Conio, M.; Bruzzi, P.; Aste, H. (1988). Family history of colorectal cancer as a risk factor for malignant tumours of the large bowel. A case control study, Int.J. Cancer, vol. 41, Nº 4, pp. 513-517.
- Cannon-Albright, L. A.; Skolnick, M. H.; Bishop, D. T.; Lee, R. G.; Burt, R. W. (1988). Common inheritance of susceptibility to colonic adenomatous polyps and associated colorectal cancers, N.Engl.J.Med, vol. 319, Nº 9, pp. 533-537.
- Martinez, M. E.; Sampliner, R.; Marshall, J. R.; Battacharyya, A. K.; Reid, M. E.; Alberts, D. S. (2001). Adenoma characteristics as risk factors for recurrednce of advanced adenomas, Gastroenterology, vol. 120, Nº 5, pp. 1077-1083.
- Atkin, W. S.; Saunders, B. P. (2002). Surveillance guidelines after removal of colorectal adenomatous polyps, Gut, vol. 51 Suppl 5, pp. V6-V9.
- Bjork, J.; Borjesson, L.; Hertervig, E.; Lindmark, G.; Ost, A. (2003). Sporadic colorectal polyps. Updated guidelines for endoscopic surveillance. Lakartidningen, vol. 100, Nº 34, pp. 2584-2588.
- Winawer, S. J.; Zauber, A. G.; Fletcher, R. H. et al. (2006). Guidelines for colonoscopy surveillance after polypectomy: a consensus update by the US Multi-Society Task Force on Colorectal Cancer and the American Cancer Society, Gastroenterology, vol. 130, Nº 6, pp. 1872-1885.
- Schmiegel, W.; Reinacher-Schick, A.; Arnold, D. et al. (2008). Update s3-guideline”colorectal cancer”2008, Z. Gastroenterol., vol. 46, Nº 8, pp. 799-840.
- Matsuda T, Chiu HM, Sano Y et al. Surveillance colonoscopy after endoscopic treatment for colorectal neoplasia: From the standpoint of the Asia– Pacific region. Digestive Endoscopy 2016; 28: 342–347.
- Van Stolk, R.; Beck, G. J.; Baron, J. A.; Haile, R.; Summer, R. (1998). Adenoma characteristic at first colonoscopy as predictors of adenoma recurrence and characteristics at first colonoscopy as predictors of adenoma recurrence and characteristics at follow-up. The polyp prevention Study Group, Gastroenterology, vol. 115, Nº 1, pp. 13-18.
- Zauber, A.; Winawer, S.; Waye, J.; Schapiro, M.; Stewart, E. T. (1999). Long term National Polyp Study(NPS) data on post-polypectomy surveillance. Endoscopy 31, E13.
- Noshirwani, K. C.; Van Stolk, R. U.; Rybicki, L. A.; Beck, G. J. (2000). Adenoma size and number are predictive of adenoma recurrence: implications for surveillance colonoscopy, Gastrointest. Endosc, vol. 51, Nº 4 pt 1, pp. 433-437.
- Hassan C, Gimeno-García A, Kalager M et al. Systematic review with meta-analysis: the incidence of advanced neoplasia after polypectomy in patients with and without low-risk adenomas. Aliment Pharmacol Ther 2014; 39: 905–912.
- Winawer, S. J.; Zauber, A. G.; Ho, M. N. et al. Prevention of colorectal cancer by colonoscopic polypectomy. The National Polyp Study Workgroup. N Engl J Med 1993; 329:1977-81
- Lotfi, A. M.; Spencer, R. J.; Ilstrup, D. M.; Melton, L. J. (1986). Colorectal polyps and the risk of subsequent carcinoma, Mayo Clin, Proc., vol. 61, Nº 5, pp. 337-343.
- Atkin, W. S.; Morson, B. C.; Cuzick, J. (1992). Long-term risk of colorectal cancer after excision of rectosimoid adenomas, N. Engl. J. Med., vol. 326, Nº 10, pp. 658-662.
- Khoury, D. A.; Opelka, F. G.; Beck, D. A. et al. (1996). Colon surveillance after colorectal cancer surgery, Dis. Colon Rectum, vol. 39, Nº 3, pp. 252-256.
- Wegener, M.; Borsch, G.; Scmidt, G. (1986). Colorectal adenomas. Distribution, incidence of malignant transformation, and rate of recurrence, Dis. Colon Rectum, vol. 29, Nº 6, pp. 383-387.
- Kahi Ch J, Boland R, Dominitz JA et al. (2016) Colonoscopy surveillance after colorectal cancer resection: recommendations of the US multi-society task force on colorectal cancer. Gastrointestinal Rndoscopy; 83, (3). 489–498.e10.
- Schreiner MA, Weiss DG, Lieberman DA. Proximal and large non-neoplastic serrated polyps: association with synchronous neoplasia at screening colonoscopy and with interval neoplasia at follow-up colonoscopy. Gastroenterology. 2010;139 (5):1497-1502
CÁNCER COLORRECTAL HEREDOFAMILIAR
El cáncer colorrectal (CCR) es considerado una enfermedad multifactorial. Influyen en su aparición causas externas (ambientales), los hábitos de vida y genéticas (propias del individuo), que interactúan y determinan la mayor o menor expresión de la enfermedad.1,2 Debido a esto, el riesgo de padecer la enfermedad es variable en cada individuo y puede ser estratificado en distintos niveles clínicos como: población general, riesgo moderado y riesgo alto. El conjunto de rasgos y alteraciones genéticas que aumentan el riesgo de aparición de CCR (susceptibilidad genética) es muy heterogéneo y, en más de la mitad de los casos, actualmente desconocido.
La identificación y caracterización correcta del riesgo de padecer CCR es actualmente un estándar de cuidado que todo especialista debe ofrecer como parte de la práctica clínica habitual. No obstante, debido a la complejidad en la detección y manejo de los pacientes y familias con alto riesgo, la estratificación de riesgo y su evaluación debe realizarse dentro del marco de una consulta de Asesoramiento Genético en oncología, llevada a cabo por un profesional entrenado.
Estratos de riesgo
El 25-30% de los casos de CCR presentan agregación familiar, donde la predisposición a desarrollar la enfermedad se encuentra aumentada.3-5 Esta predisposición puede ser muy variable y abarcar casos donde el riesgo de desarrollar la enfermedad es muy elevado (80-100%) hasta casos donde la probabilidad es sutilmente mayor a la de la población general.6
La historia familiar de cáncer colorrectal es considerado uno de los factores de riesgo de mayor peso para el desarrollo de la enfermedad, y el riesgo
aumenta según el número de familiares afectados y la edad de aparición de la enfermedad.7
Estudios realizados en gran cantidad de individuos sobre la expresión o número de copias de todo el genoma (Genome-Wide Association Studies) han logrado identificar numerosos genes y sus variantes polimórficas, que se asocian con aumentos leves del riesgo de CCR. Estas variantes suman sus efectos cuando concurren en un mismo individuo, pudiendo aumentar el riesgo de desarrollar la enfermedad en forma significativa, según el número de variantes presentes. Aún no existen en la actualidad recomendaciones de prevención específicas dirigidas a estos casos, que justifiquen su estudio sistemático en forma asistencial.8,9
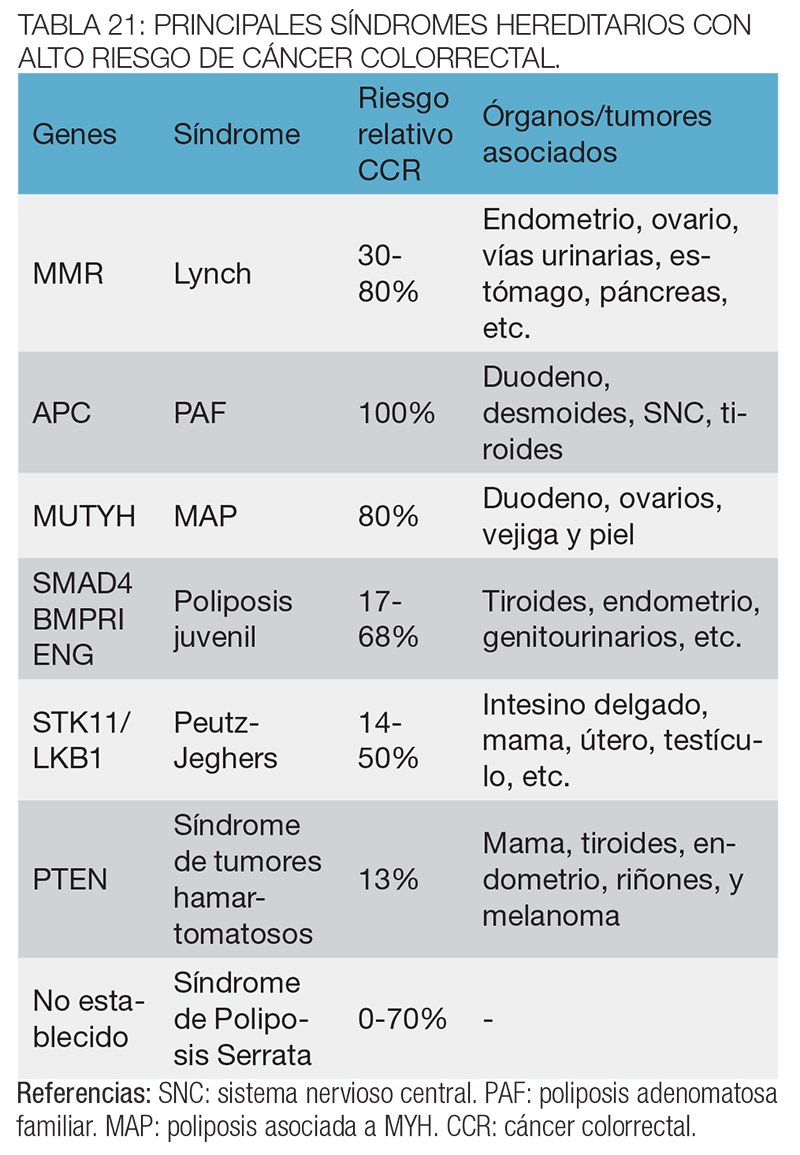
En el otro extremo del espectro se encuentran los distintos síndromes de CCR hereditario producidos por mutaciones heredables en genes puntuales, que confieren riesgos muy elevados de desarrollar la enfermedad y en general se asocian también a la aparición de tumores en otros órganos. Estos casos explican aproximadamente un 25% de los CCR familiares y cada uno de ellos posee sus propios criterios de detección, diagnóstico y prevención definidos (tabla 21).
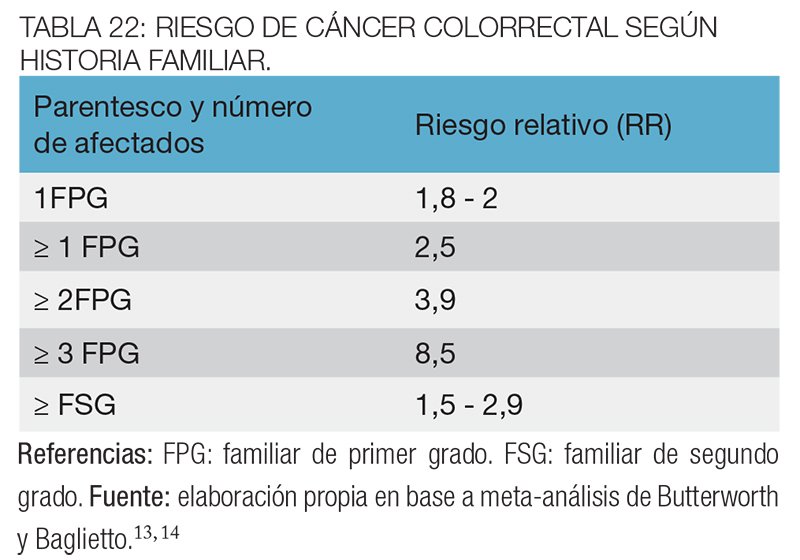
Los individuos con antecedentes familiares de CCR que no cumplen criterios específicos para sospechar ningún síndrome hereditario en relación al CCR, o que no han sido estudiados para ninguno de estos, deben igualmente ser asesorados y su riesgo puede ser calculado en forma empírica, con el objeto de adecuar las estrategias de prevención10-12 (tabla 22).

Asesoramiento genético y manejo empírico
El asesoramiento genético es el proceso mediante el cual se identifican y asesoran individuos y familias en riesgo de poseer cáncer familiar o hereditario, abarcando la complejidad de aspectos médicos, psicosociales y éticos que caracterizan a este síndrome. Este proceso forma parte del accionar de profesionales especializados en el área de la genética oncológica, trabajando en conjunto con un equipo multidisciplinario de profesionales. Además, abarca todos los pasos necesarios para la detección y para el seguimiento de estas familias. Dentro de los cuales se encuentran las características clínicas tanto individuales como personales (forma de aparición, edad de diagnóstico y agregación familiar de CCR), que permiten orientar el caso en estudio y son herramientas fundamentales para la derivación de pacientes hacia una consulta de evaluación y caracterización más exacta del riesgo (recomendación A).
En la tabla 4 se describen los principales hallazgos clínicos que presentan los CCR hereditarios, que permiten diferenciarlos de los esporádicos y son a la vez elementos fundamentales de sospecha y derivación, para profundizar el estudio individual y familiar del caso.15,16 Estas son pautas de alarma importantes para todo profesional dedicado al manejo del CCR en cualquiera de sus aspectos. Todo individuo que presente alguna de las siguientes características clínicas se beneficiaría de una consulta de asesoramiento genético oncológico, donde se evalúe el caso y la necesidad de avanzar con estudios moleculares dirigidos a algún síndrome en particular.
Cada uno de los síndromes hereditarios previamente descriptos posee criterios clínicos diagnósticos definidos, que orientan al diagnóstico molecular. Además, cada gen involucrado requiere técnicas específicas de estudio y algoritmos diagnósticos que deben ser pedidos e interpretados por profesionales entrenados en estas patologías.17
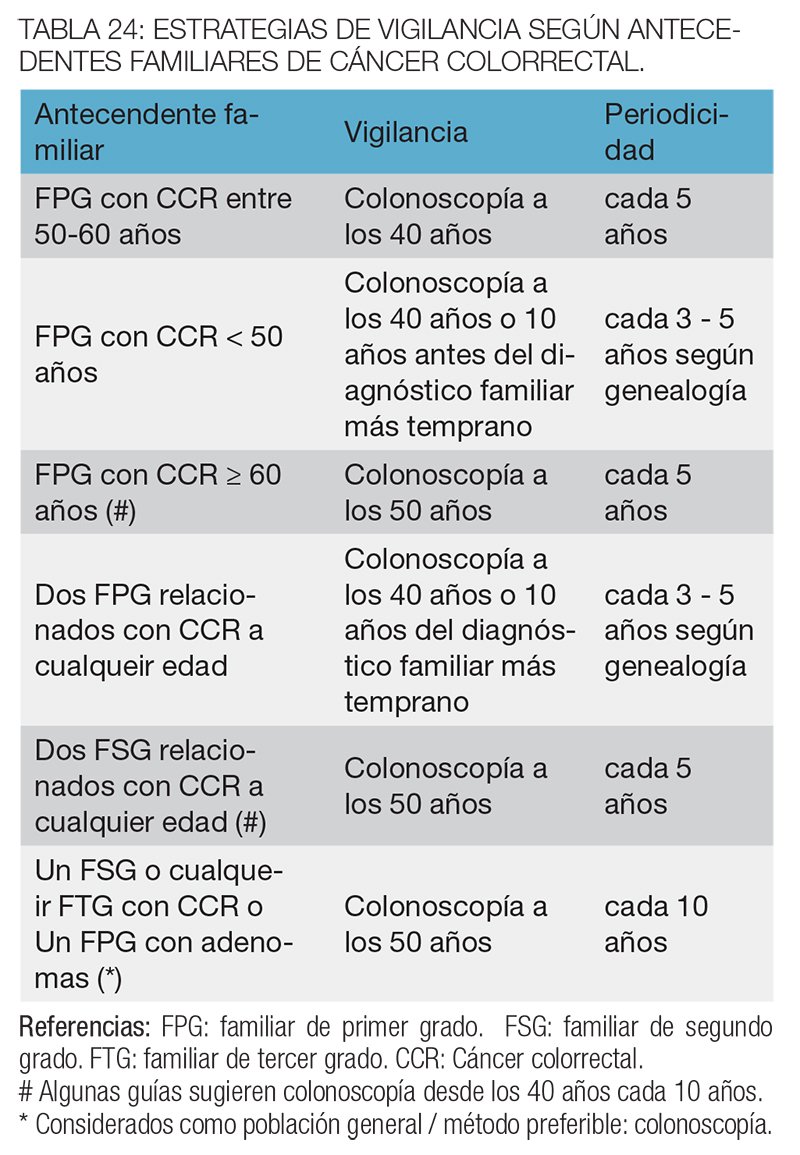
En aquellos casos donde no se encuentra un diagnóstico de un síndrome especifico, es posible aplicar estrategias de prevención empíricas, de acuerdo a los casos aparecidos en la genealogía. A continuación se resumen las estrategias sugeridas por la mayoría de las guías internacionales18 (recomendación B) (tabla 24).
BIBLIOGRAFÍA
- Daley, D.; Lewis, S.; Platzer, P. et al. (2008): “Identification of susceptibility genes for cancer in a genome-wide scan: results from the colon neoplasia sibling study”. En: Am J Hum Genet, 82(3): 723-736.
- Dermitzakis, E.T. y Clark, A.G. (2009): “Genetics. Life after GWA studies”. En: Science, 326(5950): 239-240.
- Mitchell. R.J.; Campbell, H.; Farrington, S.M. et al. (2005): “Prevalence of family history of colorectal cancer in the general population”. En: Br J Surg, 92: 1161-1164.
- Stephenson, B.M.; Finan, P.J.; Gascoyne, J. et al. (1991): “Frequency of familial colorectal cancer”. En: Br J Surg, 78: 1162-1166.
- Slattery, M.L.; Kerber, R.A. (1994): “Family history of cancer and colon cancer risk: the Utah Population Database”. En: J Natl Cancer Inst., 86: 1618-1626.
- Fuchs, C.S.; Giovannucci, E.L.; Colditz, G.A. et al. (1994): “A prospective study of family history and the risk of colorectal cancer”. En: N Engl J Med, 331(25): 1669-1674.
- Johns, L.E. y Houlston, R.S. (2001): “A systematic review and meta-analysis of familial colorectal cancer risk”. En: Am J Gastroenterol. 96: 2992-3003.
- Tenesa, A. y Dunlop, M.G. (2009): “New insights into the aetiology of colorectal cancer from genome-wide association studies”. En: Nat Rev Genet, 10(6): 353-358.
- Webb, E.; Broderick, P.; Lubbe, S.; Chandler, I.; Tomlinson, I. y Houlston, R.S. (2009): “A genome-wide scan of 10 000 gene-centric variants and colorectal cancer risk”. En: Eur J Hum Genet, 17(11): 1507-1514.
- Dunlop, M.G. y Campbell, H. (1997): “Screening for people with a family history of colorectal cancer”. En: BMJ, 314: 1779-1780.
- Burt, R.W. (2000): “Colon cancer screening”. En: Gastroenterology, 119(3): 837-853.
- Winawer, S.; Fletcher, R.; Rex, D. et al. (2003): “Colorectal cancer screening and surveillance: clinical guidelines and rationale-update based on new evidence”. En: Gastroenterology, 124(2): 544-560.
- Baglieto, L.; Jenkins, M.; Severi, G.; Giles, G.; Bishop, D.T.; Boyle, P. et al. (2006): “Measures of familial aggregation depend on the definition of family history: Meta-analysis for colorectal cancer”. En: J Clin Epidemiol., 59: 114-124.
- Butterworth, A.S.; Higgins, J.P.T. y Pharoah, P. (2006): “Relative and absolute risk of colorectal cancer for individuals with a family history. A meta-analysis”. En: Eur J Cancer, 42: 216-227.
- Grady, W.M. (2003): “Genetic testing for high-risk colon cancer patients”. En: Gastroenterology, 124: 1574-1594.
- Houlston, R.S.; Murday, V.; Harocopos, C. et al. (1990): “Screening and genetic counselling for relatives of patients with colorectal cancer in a family cancer clinic”. En: BMJ, 301: 366-368
- NCCN (s/f): “Guidelines Genetic/Familial High Risk assessment: Colorectal”. Disponible online en: <https://www.nccn.org>.
- Dove-Edwin, I. Sasieni, P.; Adams, J. y Thomas, H.J. (2005): “Prevention of colorectal cancer by colonoscopic surveillance in individuals with a family history of colorectal cancer: 16 year, prospective, follow-up study”. En: BMJ, 331: 1047.
SÍNDROME DE LYNCH
El síndrome de Lynch, previamente denominado “cáncer colorrectal hereditario no polipósico”, es una enfermedad hereditaria con patrón autosómica dominante, y es el CCR hereditario más frecuente,1 siendo responsable del 2-4% de todos los casos de CCR.2 Está asociado a mutaciones germinales en algún gen reparador del ADN ó mismatch repair genes (MMR): MLH1 y MSH2 (80%), MSH6 (10-12%), y PMS2 (2-3%). Estos genes reparan los errores de apareamiento de bases nucleotídicas ocurridos normalmente durante la replicación del ADN; su mutación determina un estado de inestabilidad de microsatélites (IMS) y la pérdida de expresión de la proteína correspondiente al gen mutado, considerándose ambas alteraciones marcadores fenotípicos del síndrome.3
Generalidades del síndrome de Lynch
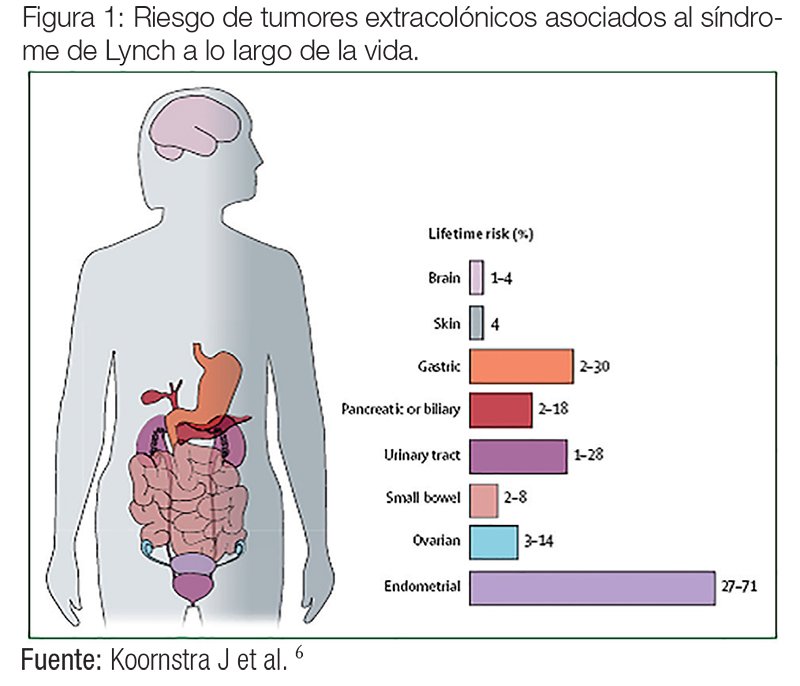
Un individuo portador de una mutación en uno de estos genes tiene un riesgo acumulado a lo largo de su vida del 30-70% para desarrollar CCR, 30-70% para cáncer de endometrio, 10-15% para tumores de ovario o estómago, y un riesgo superior a la población general para tumores de vías urinarias, intestino delgado, vía biliar, cerebro (glioblastomas), páncreas y tumores cutáneos (queratoacantomas, adenomas o adenocarcinomas sebáceos de la piel) (figura 1).4-6
Se ha observado una correlación genotipo-fenotipo, en la cual los portadores de mutaciones en el MLH1 poseen mayor riesgo de CCR a edades jóvenes, en el MSH2 poseen mayor riesgo de tumores extracolónicos, en el MSH6 poseen mayor riesgo de cáncer de endometrio, y en el PMS2 presentan un menor riesgo absoluto de CCR y de cáncer de endometrio (15-20%).5
Las principales características clínicas de esta entidad son: diagnóstico del CCR a edades tempranas (promedio 45 años de edad); la afectación predominante del colon derecho (en el 70% de los casos); la alta incidencia de tumores colorrectales sincrónicos (10%) y metacrónicos (40%), la asociación con los tumores extracolónicos ya mencionados, y la presencia de tumores con histología sugestiva de IMS: carcinoma mucinoso, con células en anillo de sello, crecimiento medular, bajo grado de diferenciación celular, infiltración linfocitaria y/o con reacción Crohn-like.6
Identificación de individuos con riesgo de padecer Síndrome de Lynch
El síndrome de Lynch no tiene un fenotipo claro que facilite la sospecha diagnóstica, por lo que se han desarrollado criterios clínicos basados en la historia personal y familiar de tumores para identificar las familias con probable síndrome de Lynch. En 1989 el International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer propuso los criterios de Ámsterdam I para unificar las características clínicas del síndrome de Lynch 7 siendo estos modificados posteriormente para incluir el riesgo aumentado de tumores extracolónicos (criterios de Ámsterdam II, tabla 25).8
La baja sensibilidad de los criterios de Ámsterdam y su limitada aplicabilidad a la práctica clínica, ha llevado a establecer criterios menos restrictivos que permiten identificar a una mayor proporción de individuos con síndrome de Lynch. Estos criterios fueron elaborados por un grupo de trabajo del National Cancer Institute en Bethesda9, y posteriormente revisados, modificados y publicados en el 2004.10 Son llamados “Criterios de Bethesda” (tabla 26) y se utilizan actualmente para identificar individuos con CCR en quienes deben realizarse los estudios moleculares específicos para confirmar o descartar la sospecha clínica de síndrome de Lynch. Además, en el último algoritmo diagnóstico del síndrome se agregó otro criterio de sospecha: mujeres con cáncer de endometrio antes de los 50 años de edad.3
Cribado molecular del síndrome de Lynch
Una vez que se identifica un individuo con CCR y criterios de Bethesda, la sospecha de padecer un síndrome de Lynch debe confirmarse mediante la identificación de una mutación germinal en algún gen reparador del ADN. El estudio genético implica tecnología molecular que es altamente costosa y de disponibilidad limitada, por lo que, una vez que se identifican individuos con criterios de Ámsterdam, individuos con uno o más criterios de Bethesda, o mujeres con cáncer de endometrio antes de los 50 años de edad, se realizan estudios moleculares a modo de rastreo (screening o cribado molecular) para identificar un déficit del sistema reparador del ADN (SRA), y así seleccionar a aquellos individuos con sospecha clínica de síndrome de Lynch que deberían someterse a un estudio genético definitivo en su ADN.11
Existen dos métodos de cribado molecular disponibles para identificar un CCR con déficit del SRA: el estudio de IMS, y el estudio de la expresión inmunohistoquímica (IHQ) de las proteínas reparadoras del ADN. 12

El estudio de IMS posee una sensibilidad del 90-95% para detectar individuos con síndrome de Lynch. El mismo se realiza comparando ADN tumoral vs. ADN normal (de tejido colónico adyacente o de leucocitos de sangre periférica), sobre un panel de 5 marcadores microsatelitales recomendados por el Panel de Bethesda.
Cuando existen cambios en 2 o más marcadores en el ADN tumoral comparado con el ADN normal, el tumor se clasifica como IMS; cuando existen cambios en 1 microsatelite o cuando no hay cambios en ninguno de los 5 marcadores se clasifica al tumor como MSS (estabilidad microsatelital). Muchos de los CCR consecuencia de una mutación germinal en el gen MSH6 evidencian MSS.13
El estudio IHQ de las proteínas reparadoras del ADN posee una sensibilidad del 90-95% para síndrome de Lynch, y se refiere a la tinción inmunohistoquímica de células del tejido tumoral en búsqueda de la expresión de las cuatro proteínas codificadas por los genes reparadores del ADN.
Un estudio IHQ normal es aquel en el cual las cuatro proteínas se expresan normalmente en el tejido tumoral en comparación con el tejido colónico normal adyacente. Un estudio IHQ anormal es aquel en el cual existe un déficit de expresión de alguna de las cuatro proteínas en el tejido de CCR en comparación con la tinción en la mucosa normal adyacente, y dirige el estudio genético al gen correspondiente.14
Las proteínas reparadoras del ADN suelen actuar en dímeros (MLH1 con PMS2 y MSH2 con MSH6); por lo tanto, si existe un déficit de expresión de MLH1/PMS2, se debe sospechar una mutación en el gen MLH1, y si existe un déficit de expresión de MSH2/MSH6, se debe sospechar una mutación en el gen MSH2. En cambio, si existe un déficit aislado de una sola proteína, se debe sospechar una mutación en el gen correspondiente.15

Sin embargo, es importante recalcar como ya se ha mencionado que el 10-15% de los CCR esporádicos presentan IMS debido a la hipermetilación de la región promotora del gen MLH1, generalmente consecuencia de una mutación somática del gen BRAF (la mutación V600E). Por lo tanto, cuando existe déficit de expresión de MLH1 en la IHQ, antes de estudiar el gen MLH debe descartarse tal mutación y/o la hipermetilación del MLH1.13
Cuando un CCR presenta IMS y/o déficit de alguna proteína reparadora del ADN en la IHQ, se considera que ese CCR posee un déficit del sistema reparador del ADN (SRA) y es definido como “dSRA” (déficit del sistema reparador del ADN). En cambio, los tumores con MSS y expresión conservada de las 4 proteínas en la IHQ presentan el sistema reparador del ADN intacto y son definidos como “iSRA”.16
Ninguno de los dos métodos de cribado molecular posee una sensibilidad del 100% para detectar tumores con dSRA, por lo que son complementarios. En el 5-8% de los casos, existe IMS con expresión conservada de las 4 proteínas, o MSS con déficit de expresión de alguna de las proteínas reparadoras.4 Por lo tanto, si se realizan los dos estudios moleculares de rastreo, se aumenta al 98-100% la probabilidad de identificar individuos con riesgo síndrome de Lynch.
Algoritmo diagnóstico molecular del síndrome de Lynch (gráfico 28):17
- Si existe expresión conservada de las cuatro proteínas reparadoras en el tejido tumoral, se debe complementar con el estudio de IMS:
- Si el estudio de IMS clasifica el tumor como MSS, entonces se descarta el síndrome de Lynch, y se clasifica el CCR como esporádico.
- Si el estudio de IMS clasifica el tumor como MSI, entonces se sospecha una mutación germinal en alguno de los 4 genes reparadores (con IHQ normal), y se debe realizar el estudio genético de los genes MLH1 y MSH2. Si no se encontrara ninguna mutación germinal en estos dos genes, se debería completar el análisis con el estudio genético de los genes MSH6 y PMS2.
- Si existe déficit de expresión de alguna de las proteínas reparadoras en el tejido tumoral, la conducta a seguir depende de la proteína ausente:
- Si existe d éficit de expresión de MLH1/PMS2 o de MLH1 aislada, debe realizarse el estudio de la mutación V600E del gen BRAF y/o el estudio de metilación de la región promotora del gen MLH1. Si el estudio de mutación V600E del gen BRAF y/o el estudio de metilación de la región promotora del gen MLH1 es/son positivos, se confirma el CCR como esporádico con IMS (descartándose un síndrome de Lynch). En cambio, si ambos estudios son negativos se descarta el CCR esporádico y debe analizarse el gen MLH1.
- Si existe déficit de expresión de PMS2, debe realizarse el estudio genético del gen PMS2.
- Si existe déficit de expresión de MSH2/MSH6 o déficit de MSH2 aislada, debe realizarse el estudio genético del gen MSH2.
- Si existe déficit de expresión de MSH6 aislada, debe realizarse el estudio genético del gen MSH6.
Estudio genético en el síndrome de Lynch
El estudio de las mutaciones responsables del síndrome de Lynch se realiza en ADN del individuo afectado. Estas mutaciones se presentan en forma heterozigota en la mayoría de los casos. El 70% de las mismas implican pequeñas mutaciones que afectan pocos nucleótidos (cambios en un nucleótido, pequeñas inserciones o deleciones) y son detectadas mediante la ampliación y secuenciación por PCR de las regiones codificantes (exones) y de las regiones vecinas de los exones de los genes implicados.18 Sin embargo, en casi un tercio de los pacientes con síndrome de Lynch existen grandes deleciones o reordenamientos en algún gen reparador del ADN que no pueden ser detectadas cuando se realiza el rastreo de mutaciones puntuales por PCR y secuenciación. Para detectar este tipo de mutaciones es necesario el empleo del método MLPA (Multiplex Ligation-dependent Probe Amplification) seguido de electroforesis capilar para su detección.19 Así, el estudio genético germinal completo del síndrome de Lynch incluye tanto la secuenciación del ADN como el análisis con MLPA.4 Sin embargo, aún cuando se utilizan las dos técnicas complementarias, la tasa de detección de mutaciones germinales patogénicas (causantes de la enfermedad) en individuos con dSRA es del 30-70%.20
Cuando se identificaron por primera vez los genes responsables del síndrome de Lynch en 1992, las estrategias de secuenciación detectaban menos de la mitad de las mutaciones identificadas hoy en día con la nueva tecnología disponible.21 Desde entonces y hasta ahora se pensaba que todos los casos con dSRA sin metilación del MLH1 eran consecuencia de un síndrome de Lynch, aún en el 30-70% de los casos en los cuales no se podía identificar una mutación en algún gen reparador del ADN. Así, se denominaba “probable síndrome de Lynch” a los individuos con dSRA en el CCR y test genético negativo. Un artículo muy reciente del mes de mayo de este año Rodriguez-Soler et al.22 sugiere que podría haber más en esta historia.
Los autores analizaron 1705 individuos con CCR incluidos en 2 estudios multicéntricos poblacionales durante el 2000-2001 (EPICOLON I)23 y 2006-2007 (EPICOLON II).24 Realizaron los estudios de IMS e IHQ en todas las muestras de CCR, y seleccionaron los casos con IMS, déficit de expresión de alguna proteína en la IHQ y estudio de metilación del MLH1 negativo. Analizaron luego en estos casos los genes reparadores del ADN correspondientes según la pérdida de la expresión proteica en la IHQ.
Identificaron 56 casos con dSRA y metilación del MLH1 negativa (56/1705, 3.2% de la cohorte total), pero encontraron una mutación patogénica en algún gen reparador en solo 16 (16/1705, 0.9% de la cohorte total). Por lo tanto, la tasa de detección de una mutación germinal en individuos con dSRA y metilación del MLH1 negativa fue del 29% (16/56). A los 40 individuos con dSRA y estudio genético negativo (40/56, 71%) los denominaron “Lynch-like syndrome” o síndrome de Lynch-símil (SLS).
Luego compararon el riesgo de CCR, el riesgo de tumores extracolónicos, y la edad diagnóstica del CCR en familiares de primer grado (1°) de los individuos con SLS vs. los familiares 1° de los individuos con síndrome de Lynch confirmado y los familiares de los individuos con CCR esporádico (iSRA). Este análisis no había sido realizado previamente en la literatura médica.

Evidenciaron que la incidencia de CCR en los familiares de los individuos con SLS fue significativamente más baja que en los familiares de los individuos con síndrome de Lynch confirmado (probabilidad estandarizada de incidencia -SIR- de 2.12 vs. 6.04 respectivamente, p<0.001), y significativamente más alta que en los familiares de los individuos con CCR esporádico (SIR 2.12 vs. 0.48 respectivamente, p<0.001). El SIR para tumores extracolónicos fue más alto, aunque sin significación estadística, en los familiares de individuos con síndromes de Lynch (SIR 2.81) en comparación con los familiares de los individuos con SLS (SIR 1.69, p 0.09) y con los familiares de los CCR esporádicos (1.2, p 0.5).
Con respecto a la edad diagnóstica del CCR en los familiares, los familiares de 1° de los individuos con SLS desarrollaron la enfermedad a una edad similar que la de los familiares de los individuos con síndrome de Lynch (53.7 vs. 48.5, p 0.23), y a una edad más temprana que los familiares de los individuos con CCR esporádico (53.7 vs. 68.8, p 0.004).22
El trabajo de Rodriguez-Soler y col.22 plantea el nuevo concepto de SLS: casos de CCR con dSRA y metilación negativa/BRAF del MLH1 que no son síndrome de Lynch. Probablemente algunos estos sean verdaderos casos de síndrome de Lynch en los cuales no se ha podido identificar una mutación germinal en algún gen reparador mediante el conocimiento y la tecnología disponible en la actualidad. Sin embargo, las diferencias evidenciadas en los familiares de 1° de los individuos con SLS sugieren que podría existir otro mecanismo molecular, todavía no descubierto, generador de un déficit del SRA distinto a una mutación en algún gen reparador del ADN.21
Aunque el mecanismo para la generación del dSRA en este escenario es desconocido, se plantean tres probables alternativas: que existan mutaciones crípticas en alguno de los genes reparadores del ADN (o sea, existe una mutación patogénica pero esta no es identificada mediante el conocimiento y la tecnología disponible en la actualidad), que exista un proceso somático en los genes reparadores del ADN que generen dSRA mediante mutaciones bialélicas somáticas de estos genes, y/o que existan otros genes no descubiertos hasta el momento (distintos a los reparadores del ADN) que generen dSRA.25
Con respecto a la segunda posibilidad, existen tres estudios recientes que han analizado este mecanismo con 23,26 1827 y 3528 casos de CCR con SLS. Ellos identificaron una inactivación somática bialélica del gen MLH1 o MSH2 en el 52, 69 y 65% de los casos de SLS respectivamente, y plantearon que este podría ser el mecanismo molecular subyacente en más de la mitad de los casos de SLS, aunque no pueden descartar que además exista en este escenario una mutación a nivel germinal.
Por último, existe la tercera alternativa: la presencia de genes con predisposición a CCR no descubiertos que generen dSRA. Esta posibilidad puede ser explorada mediante el estudio de exomas germinales en estos pacientes.25 No hemos identificado en la bibliografía un estudio de este tipo en línea germinal en individuos con síndrome de Lynch-símil.
Consejo genético en familiares de individuos con síndrome de Lynch y SLS
Una vez realizados los estudios moleculares correspondientes en un individuo con CCR y sospecha de síndrome de Lynch, debería aconsejarse a sus familiares de 1° la realización de una consulta de asesoramiento genético. En esta, si se logró identificar una mutación germinal patogénica en el individuo afectado, se debe ofrecer un estudio genético dirigido para identificar quienes heredaron la mutación (portadores sanos) y quienes no, y según los resultados adecuar las conductas de vigilancia. Además, también se deben indicar las conductas de vigilancia adecuadas a los familiares de individuos con SLS.29
Vigilancia del cáncer en individuos con síndrome de Lynch
La vigilancia endoscópica del CCR es la única estrategia que demostró reducir la incidencia y la mortalidad por cáncer en el síndrome de Lynch.29 La vigilancia de los otros órganos en los cuales se pueden desarrollar los tumores asociados al síndrome no ha demostrado hasta el momento una reducción de su incidencia ni de su mortalidad; todavía no está claro que tipo de tumores extracolónicos se benefician de un programa de vigilancia, a qué edad se debería comenzar, ni los intervalos de la misma.29
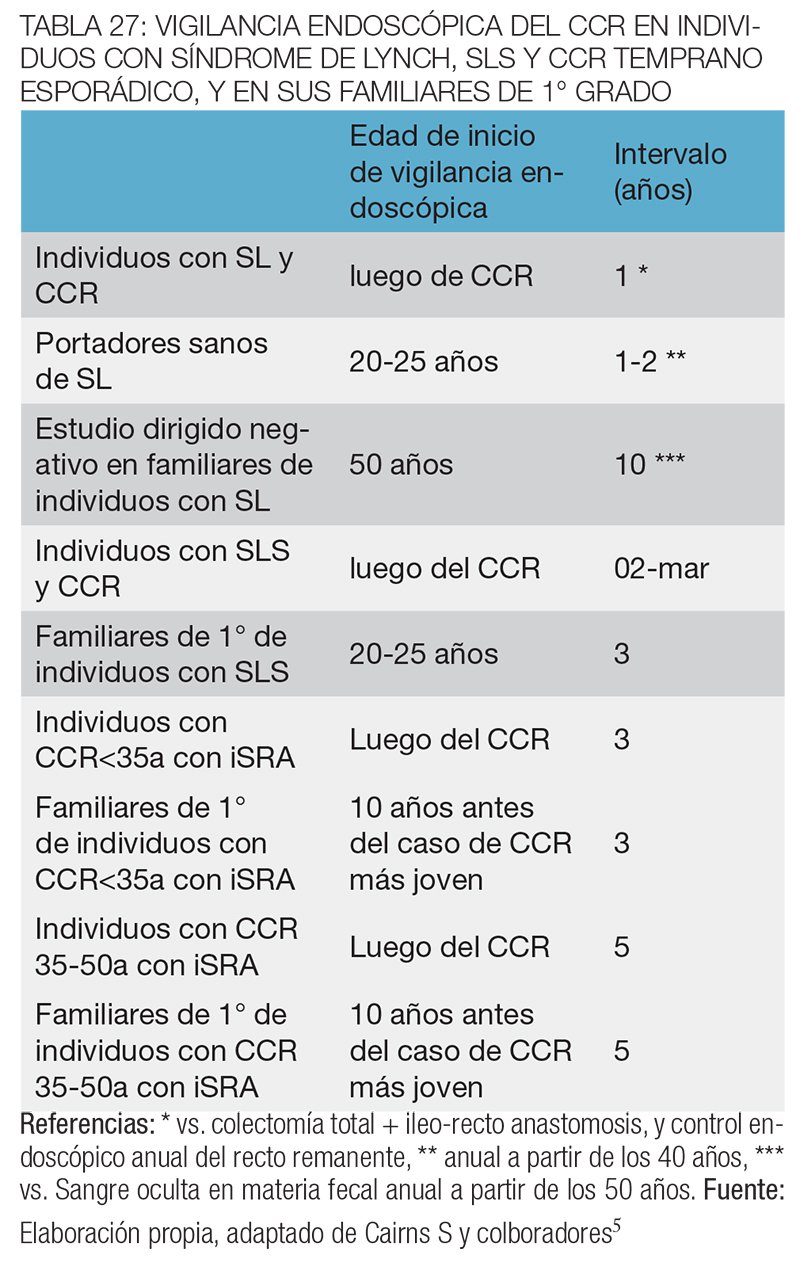
Vigilancia del CCR metacrónico en individuos con síndrome de Lynch y CCR: los individuos que hayan desarrollado CCR deben ser incluidos en un programa intensivo de vigilancia endoscópica anual del colon/recto remanente, ya que existe un riesgo elevado de padecer CCR metacrónico: un 16% y un 40% de estos individuos desarrollarán un segundo CCR dentro de los 10 y 30 años posteriores al diagnóstico inicial, respectivamente.30
Vos tot Nederveen Cappel W et al.31 compararon la expectativa de vida de individuos con síndrome de Lynch y CCR en quienes se les realizaba una colectomía total vs. una resección segmentaria + colonoscopías anuales. Los resultados indicaron que la colectomía total en pacientes menores de 47 años de edad mejoraba las expectativas de vida en 2.3 años. Por lo tanto, en pacientes con síndrome de Lynch confirmado y CCR antes de los 50 años de edad el tratamiento de elección es la colectomía subtotal. Es importante destacar que en este estudio no se comparó la calidad de vida según los tipos de cirugía.
Vigilancia del CCR en portadores sanos de síndrome de Lynch: se debe iniciar la vigilancia endoscópica a los 20-25 años de edad en todos los individuos portadores sanos de una mutación germinal en algún gen reparador del ADN. En aquellas familias en las cuales exista un individuo con CCR antes de los 25 años, se recomienda iniciar la vigilancia 2-5 años antes del diagnóstico del CCR más temprano. No hay establecido hasta la fecha un límite de edad en la cual se debería finalizar la vigilancia; este límite debe basarse en el estado de salud de cada individuo.32 El intervalo óptimo de las colonoscopías de seguimiento varía entre 1-2 años. No hay estudios que comparen los distintos intervalos; aunque un estudio finlandés evidenció que la colonoscopía cada 3 años redujo significativamente la incidencia y la mortalidad por CCR en estas familias,33 la secuencia adenoma-carcinoma está acelerada en este síndrome, y por lo tanto los expertos recomiendan que el intervalo entre colonoscopías sea de 1-2 años hasta los 40 años, y anual a partir de esa edad.34
Los familiares en quienes el estudio mutacional dirigido resulte negativo, se descarta la presencia del síndrome de Lynch, y se aconseja realizar el cribado para CCR utilizado en individuos con riesgo promedio de CCR (colonoscopías desde los 50 años de edad, y cada 10 años vs. estudio de sangre oculta en materia fecal anual a partir de los 50 años) (tabla 27).35
Vigilancia del cáncer de endometrio: en mujeres con síndrome de Lynch, el riesgo de desarrollar cáncer de endometrio es igual o mayor que el riesgo de desarrollar CCR, presentando un riesgo acumulado a lo largo de la vida del 20-70%, comparado con el 3% de la población general. En series no seleccionadas de mujeres con cáncer de endometrio, aproximadamente el 1.8% de los casos se asocian al síndrome de Lynch.36
Existen pocos estudios sobre la eficacia de la vigilancia ginecológica en mujeres con síndrome de Lynch. Dos de ellos37,38 evaluaron la utilidad de la ecografía transvaginal anual para detectar el carcinoma endometrial temprano en esta población, sin resultados positivos. En cambio, un tercer estudio evaluó la eficacia de una ecografía transvaginal + una biopsia endometrial anual y evidenció que esta medida es efectiva para la detección temprana del carcinoma endometrial en mujeres con síndrome de Lynch. Sin embargo, no demostró reducir la mortalidad por este tumor.39
Un estudio retrospectivo observacional en mujeres con síndrome de Lynch demostró la ausencia de cáncer de endometrio y de ovario en mujeres en las cuales se realizó una histerectomía + salpingooforectomía bilateral profiláctica, comparadas con una incidencia del 33% y del 5% de cáncer de endometrio y de ovario, respectivamente, en mujeres en quienes no se realizó ninguna cirugía.40 Por lo tanto, aunque las guías recomiendan una ecografía transvaginal + biopsia endometrial anual a partir de los 30-35 años de edad, la única medida que ha demostrado la reducción de la mortalidad por cáncer de endometrio en mujeres con síndrome de Lynch es la histerectomía profiláctica. Esta medida se recomienda especialmente en mujeres mayores a 45 años que hayan completado su planeamiento familiar.5
Vigilancia del cáncer gástrico: el riesgo de desarrollar cáncer gástrico en individuos con síndrome de Lynch varía ampliamente entre distintas publicaciones (2-30%).41 Se recomienda una endoscopía digestiva alta cada 1-2 años iniciando a los 30-35 años de edad, asociado a la erradicación del Helicobacter pylori, únicamente en familias con síndrome de Lynch que presenten antecedentes de cáncer gástrico.42 Sin embargo, algunos expertos recomiendan estas medidas aún cuando no existan antecedentes de este tumor.43
Vigilancia del cáncer de urotelio: los individuos con síndrome de Lynch tienen un riesgo del 12% (1-28%) de desarrollar carcinoma del tracto urinario, especialmente de la pelvis renal y el uréter, a lo largo de su vida.41 Las recomendaciones actuales difieren entre los distintos grupos: Lindor et al 42 sugiere realizar una citología urinaria anual a partir de los 30-35 años de edad en todos los individuos con síndrome de Lynch, y Vasen et al 32 sugiere una citología urinaria + una ecografía abdominal/renal anual o bianual, a partir de los 30-35 años de edad, únicamente en aquellas familias en las cuales exista el antecedente de cáncer urotelial.
Vigilancia del cáncer de ovario: el riesgo de desarrollar cáncer de ovario en mujeres con síndrome de Lynch es del 7% (3-14%), comparado con el 1.4% de la población general.44 Aunque algunos grupos recomiendan una ecografía transvaginal + CA 125 anual a todas las mujeres a partir de los 30-35 años de edad, la evidencia disponible no justifica la vigilancia de estos tumores.44 Como ya se mencionó, la salpingooforectomía bilateral profiláctica es la única medida que ha demostrado disminuir la incidencia del cáncer de ovario en mujeres con síndrome de Lynch.40
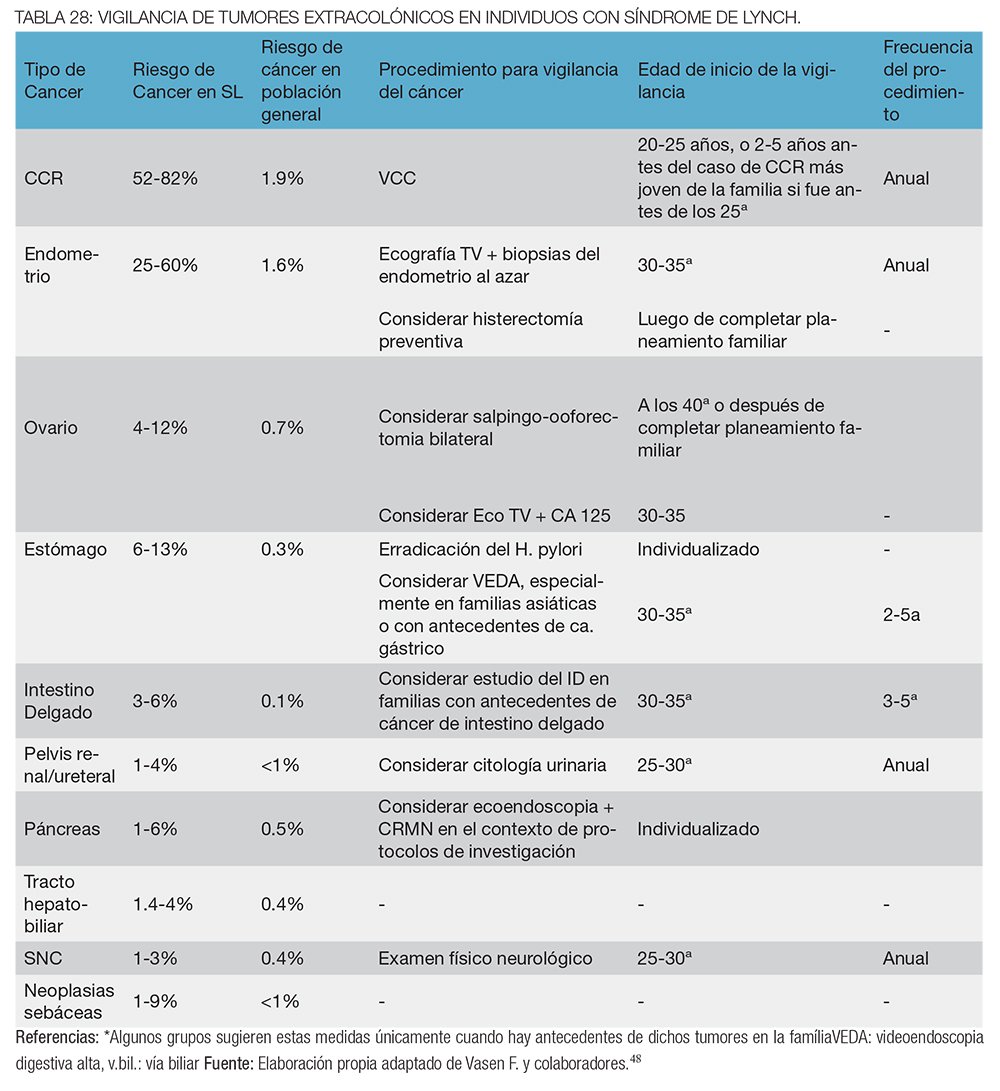
Vigilancia del cáncer de páncreas: el riesgo de padecer cáncer de páncreas en individuos con síndrome de Lynch es del 2-18%, un riesgo 7 veces mayor que el de la población general.45 Existen estudios en marcha que están evaluando la eficacia de la vigilancia con ecoendoscopía, sin resultados conocidos todavía. Por ahora, no existe ninguna recomendación para la vigilancia de estos tumores en pacientes con síndrome de Lynch.44
Vigilancia del cáncer de intestino delgado: los pacientes con síndrome de Lynch poseen un riesgo del 4% de desarrollar adenocarcinoma de intestino delgado, 100 veces superior al de la población general. Los tumores suelen presentarse en el duodeno o yeyuno, y la mayoría de ellos presentan un alto grado de IMS.45 Aunque algunos expertos32 recomiendan la vigilancia de los tumores del intestino delgado con cápsula endoscópica cada 2-3 años, iniciando a los 30-35 años de edad, todavía se necesitan más estudios que evalúen la eficacia de esta medida.
Vigilancia de los tumores de piel: la presencia de tumores sebáceos de la piel (adenomas, epiteliomas o carcinomas) y de queratoacantomas en individuos con síndrome de Lynch se denomina síndrome de Muir-Torre, y tiene una prevalencia del 10-40% en esta enfermedad.46 Estos tumores suelen aparecer en la cara, y en la mayoría de los casos, se desarrollan después del diagnóstico de otro tumor relacionado al síndrome. Se recomienda un examen dermatológico anual.32
Vigilancia de los tumores de cerebro: el riesgo de desarrollar tumores de cerebro en pacientes con síndrome de Lynch es del 2%; los tumores más frecuentes en este contexto son los glioblastomas multiformes y los astrocitomas, y cuando aparecen en pacientes con CCR se denomina “síndrome de Turcot”. Aunque presentan una baja incidencia, algunas series afirman que representan la tercera causa de muerte por cáncer en el síndrome de Lynch.47 Debido al bajo riesgo de desarrollar tumores de cerebro, a la ausencia de métodos de vigilancia para estos tumores, y a la falta de estudios que evalúen la eficacia de la vigilancia en síndrome de Lynch, no se recomienda ninguna medida hasta el momento.32
Un resumen de las recomendaciones de vigilancia de tumores extracolónicos en individuos con síndrome de Lynch se presentan el la tabla 28.
BIBLIOGRAFÍA
- Hampel H., Frankel W., Martin E. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005; 352: 1851-1860.
- Desai T., Barkel D. Syndromic Colon Cancer: Lynch Syndrome and Familial Adenomatous Polyposis. Gastroenterol Clin N Am 37 2008; 37: 47-72.
- Jenkins M., Baglietto L. Cancer risks for mismatch repair gene mutation carriers: a population-based early onset case-family study. Clin Gastroenterol Hepatol 2006; 4: 489-498.
- Boland C., Thibodeau S. A National Cancer Institute Workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998; 58: 5248-57.
- Cairns S., Scholefield J. Guidelines for colorectal cancer screening and surveillance in moderate and high risk groups (update from 2002). Gut 2010; 59: 666-689.
- Koornstra J., Mourit´s M. Management of extracolonic tumours in patients with Lynch Syndrome. Lancet Oncol 2009; 10: 400-408.
- Vasen F., Medin J. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer. Dis Colon Rectum 1991; 424.
- Vasen F., Watson P. New clinical criteria for hereditary colorectal cancer (HNPCC, Lynch Syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterol 1999; 116: 1453-1456.
- Rodriguez-Bigas M., Boland C. Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst for (1997): 1758-1762.
- Umar A., Boland C. Revised Bethesda Guidelines for Hereditary nonpolyposis colorectal cancer (Lynch Syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96: 261-268.
- Julié C., Trésallet C. Identification in daily practice of patients with Lynch Syndrome (Hereditary Nonpolyposis Coloretal Cancer): Revised Bethesda guidelines-based approach versus molecular screening. Am J Gastroenterol 2008; 103: 2825-2835.
- Hampel H., Frankel W. Screening for the Lynch Syndrome (hereditary non polyposis colorectal cancer. N Eng J Med 2005; 352: 1851-1860.
- Piñol V., Castells A. Accuracy of the revised Bethesda guidelines microsatellite instability, and immunohistochemistry for the identification of patients with hereditary non polyposis hereditary colorectal cancer. JAMA 2005; 293: 1986-1994.
- Baudhuin L., Burgart L. Use of microsatellite instability and immunohistochemistry testing for the identification of individuals at risk for Lynch syndrome. Familial Cancer 2005; 4: 255-265.
- Aarnio M., Sankila R. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 1999; 81: 214-18.
- Grady W. Genetic testing for high-risk colon cancer patients. Gastroenterology 2003; 124: 1574-1594.
- Burt R., Barthel J., Dunn K. NCCN Clinical Practice Guidelines in Oncology: Colorectal Cancer Screening 2010; 8: 8-61.
- Chao E., Velasquez J. Accurate classification of MLH1/MSH2 missense variants with multivariate analysis of protein polymorphisms-mismtach repair (MAPP-MMR). Human mutation 2008; 29: 852-860.
- Bolufer Gilabert P., Cuevas Cuerda D. Guía de práctica clínica en cáncer hereditario. Plan oncológico Comunitat Valenciana 2009: 61-77.
- Stoffel E., Chittenden A. Genetic testing for hereditary colorectal cancer: challenges in identifying, counseling, and managing high-risk patients. Gastroenterol 2010; 139: 1436-1441.
- Boland R. The mystery of mismatch repair deficiency: Lynch or Lynch-like? Gastreonterol 2013; 144: 868-881.
- Rodriguez-Soler M., Pérez-Carbonell L., Guarinos C. Risk of cancer in cases of suspected Lynch syndrome without germline mutation. Gastroenterol 2013; 144: 868-881.
- Piñol V., Andreu M., Castells A. Frequency of hereditary non-polyposis colorectal cancer and other colorectal cancer familial forms in Spain: a multicentre, prospective, nationwide study. Eur J Gastroenterol Hepatol 2004; 16: 39-45.
- Piñol V., Castells A., Andreu M. Gastrointestinal Oncology Group of the Spanish Gastroenterological Association. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA 2005; 293:1986-1994.
- Carethers J. Differentiating Lynch-like from Lynch syndrome. Gastroenterology 2014; 146: 602–614.
- Mesenkamp AR, Vogelaar IP, van Zelst-Stams WAG, et al. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch-syndrome like tumors. Gastroenterology 2014; 146: 643-646.
- Hampel H, Tomsic J. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations Gastroenterology. 2014; 147(6): 1308-1316.28. Geurts-Giele W, Leenen C, Dubbink H. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. The Journal of Pathology 2014; 234 (4): 548–559.29. Vasen H., Boland C. Progress in genetic testing, classification, and identification of Lynch syndrome. JAMA 2005; 293: 2028-2030.
- Vos tot Nederveen Cappel W., Nagengast F. Surveillance for hereditary nonpolyposis colorectal cancer: a long-term study on 114 families. Dis Colon Rectum 2002; 45: 1588-1594.
- Vos tot Nederveen Cappel W., Buskens E. Decision analysis in the surgical treatment of colorectal cancer due to a mismatch repair gene defect. Gut 2003; 52: 1752-1755.
- Vasen H., Moslein G. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis colorectal cancer). J Med Genet 2007; 44: 353-362.
- Jarvinen H., Aarnio M. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterol 2000; 118: 829-834.
- Jong A., Hendriks Y. Decrease in mortality in Lynch Syndrome families because of surveillance. Gastroenterol 2006; 130: 665-671.
- 35. Win A., Young J., Lindor N. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol 2012; 30: 958-964.
- Vasen H., Watson P. The epidemiology of endometrial cancer in hereditary nonpolyposis colorectal cancer. Anticancer Res 1994; 14: 1675-1678.
- Dove-Edwin I., Boks D. The outcome of the endometrial carcinoma surveillance by ultrasound scan in women at risk of hereditary nonpolyposis colorectal carcinoma and familial colorectal carcinoma. Cancer 2002; 94: 1708-1712.
- Rijcken F., Mourits M. Gynecologic surveillance in hereditary nonpolyposis colorectal cancer. Gynecol Oncol 2003: 74-80.
- Renkonen-Sinisalo L., Butzow R. Surveillance for endometrial cancer in hereditary nonpolyposis colorectal cancer syndrome. Int J Cancer 2007; 120: 821-824.
- Schmeler K., Lynch H. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med 2006; 354: 261-269.
- De Jong A., Hendriks Y. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology 2006; 130: 665-671.
- Aarnio M., Mecklin J. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer 1995; 64: 430-433.
- Park Y., Shin K. Risk of gastric cancer in hereditary nonpolyposis colorectal cancer in Korea. Clin Cancer Res 2000; 6: 2994-2998.
- Lindor N., Petersen G. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 2006; 296: 1507-1517.
- Watson P., Vasen H. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J cancer 2008; 123: 444-449.
- Geary J., Sasieni P. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer (HNPCC). Fam Cancer 2008; 7: 163-172.
- South C., Hampel H. The frequency of Muirr-Torre syndrome among Lynch syndrome families. J Natl Cancer 2008; 100: 277-281.
- Vasen F., Blanco I., Aktan-Collan K. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendatios by a group of European experts. Gut 2013; 0: 1-13.
POLIPOSIS ADENOMATOSA FAMILIAR Y OTRAS POLIPOSIS COLÓNICAS
Como se ha consignado en capítulos anteriores, el cáncer colorrectal (CCR) en más del 70% de los casos es esporádico. Solo un pequeño porcentaje (entre un 5 y 15%) es atribuible a síndromes hereditarios, siendo el más frecuente el cáncer colorrectal hereditario no asociado a poliposis (Síndrome de Lynch).1-6
Dentro de los síndromes de poliposis, el más frecuente es la Poliposis adenomatosa familiar (PAF), con una prevalencia de 1:6.850 a 1:31.250 nacidos vivos, con un riesgo de CCR del 100% si los pacientes no son diagnosticados y operados a tiempo.
De acuerdo a su histología, los pólipos se clasifican en:
- Síndromes de poliposis adenomatosas: Poliposis Adenomatosa Familiar (PAF) con sus variantes fenotípicas y la Poliposis Asociada a MUTHY (antes MYH) o MAP, Poliposis asociada a corrección de lectura de polimerasa (PPAP), actualmente denominadas Poliposis asociadas a POLE o POLD1, Poliposis adenomatosa asociada a mutaciones bialélicas en los genes reparadores de daño del ADN (MMR), también denominada deficiencia constitucional de los genes reparadores de daños.
- Síndromes de poliposis hamartomatosas: Síndrome de Peutz Jeghers, Síndrome de Poliposis Juvenil y Síndrome de Cowden.
- Síndromes de poliposis mixta hereditario.
- Síndrome de poliposis Serrata o Aserrada.
Poliposis Adenomatosa Familiar
La Poliposis Adenomatosa Familiar es una enfermedad hereditaria que se produce por mutaciones germinales en el gen APC (brazo largo del cromosoma 5) y se transmite en forma autosómica dominante.7,11
Se caracteriza por la presencia de decenas a miles de pólipos adenomatosos colorrectales,12 que presentan diversos fenotipos de acuerdo al número de adenomas. Se denomina poliposis atenuada aquella que presenta entre 20 y 99 adenomas, la forma clásica con cientos de pólipos pero en número menor a 1000 y las formas floridas o severas con más de 1000 pólipos o con una afectación llamada en alfombra.
Estos pólipos, siguiendo la secuencia adenoma-carcinoma, hacen que esta enfermedad tenga un riesgo de malignizacion cercano al 100%, de no mediar un diagnóstico y tratamiento temprano.5,7-9,13
Si bien la mutación responsable de la enfermedad se encuentra presente desde la concepción, los adenomas colorrectales suelen aparecer en la adolescencia, siendo excepcional la presencia de adenomas avanzados en menores de 10 años. La edad promedio de aparición del CCR es entre los 35 y los 39 años.
La afectación colorrectal se da en el 100% de los casos, siendo asintomática u oligosintomática en su inicio. La triada sintomática más frecuente es la alteración del ritmo evacuatorio, con tendencia a la diarrea, la proctorragia y la mucorrea.
Debido a su carácter germinal, los pacientes podrán desarrollar distintas manifestaciones extracolónicas, algunas de las cuales, por el riesgo de transformación hacia carcinoma o por su propio comportamiento maligno, deberán ser buscadas (adenomas duodenales, cáncer de tiroides y tumores desmoides) y otras menos frecuentes permanecen aún sin indicación formal de pesquisa (hígado 1% y cerebro < 1%).
La asociación de pólipos colorrectales con lesiones en otros órganos son variantes fenotípicas de la misma enfermedad, llevando el nombre de quien las describiera: el Síndrome de Turcot (adenomas colorrectales y tumores del SNC, generalmente gliomas malignos o meduloblastomas) y el Síndrome de Gardner (adenomas y múltiples manifestaciones extracolónicas a nivel de los tejidos blandos y duros: osteomas, adenomas duodenales, lipomas, quistes epidermoideos, hipertrofia del epitelio pigmentario de la retina).14
Si bien la PAF se produce por alteraciones genéticas, estas pueden no ser halladas hasta en un 20% de los pacientes con diagnóstico clínico y anatomopatológico. En las últimas décadas se han comenzado a encontrar otros genes responsables de poliposis colónica indistinguibles clínicamente de la PAF producida por mutaciones en el gen APC.
La PAF representa un modelo de prevención del CCR en grupos de riesgo alto, donde la pesquisa en familiares de primer grado de las personas afectadas permite detectar posibles portadores diagnosticándolos en etapa pre neoplásica, disminuyendo la incidencia de CCR o en etapas tempranas del cáncer, con mayor curación. Es una recomendación fuerte que estas familias sean controladas en el contexto de un registro de cáncer familiar, que son grupos interdisciplinarios especializados en el manejo de pacientes donde el objetivo fundamental no es solo registrarlos en una base de datos, sino una búsqueda, citación activa y seguimiento de todos los familiares afectados o en riesgo.3,7,15-20
Manifestaciones extracolónicas
Afectación del tracto digestivo alto: la afectación del tracto digestivo alto en la PAF es frecuente. Se ha descripto que entre el 50 y el 90% de estos pacientes presentan adenomas en el duodeno. El riesgo de cáncer periampular también se encuentra aumentado con respecto a la población general, con una incidencia en este grupo del 3 al 5%.
La pesquisa se realiza con endoscopias altas, con endoscopio de visión frontal y lateral, a partir de los 20-25 años, continuando el seguimiento según los hallazgos. Para ello se utiliza el score de Spiegelman, en el cual toman en cuenta el número de pólipos, el tamaño, la histología y el grado de displasia (tablas 29 y 30).7,21-22
Se sugiere realizar tratamiento endoscópico siempre que sea posible, con resección de todas las lesiones mayores a 1cm. Los pacientes que presenten un Spiegelman 4 deberán ser evaluados en equipos multidisciplinarios para determinar la conducta a seguir. En aquellos con afectación duodenal severa y sospecha de malignidad o presencia de cáncer, la duodenopancreatectomia cefálica con gastroyeyunoanastomosis se encuentra indicada.23 En aquellos casos donde se descarta malignidad, la duodenectomía con preservación pancreática es una opción aceptada.24 Las resecciones menores, si bien son factibles, tienen altas tasas de recidiva.
Diversos estudios han demostrado que tanto el uso de Sulindac como el de Celecoxib a dosis de 800 mg/día disminuyen el número y tamaño de los pólipos durante el tratamiento, reapareciendo ante la suspensión. La utilización de estas drogas en nuestro país es poco viable debido la falta de disponibilidad.
Si bien no es común la presencia de adenomas en el intestino delgado (más frecuente cuando hay afectación duodenal severa), en esos casos, se encuentran indicados estudios del intestino delgado (cápsula endoscópica, entero resonancia, entero tomografía o tránsito de intestino delgado).25
Se han descripto casos de adenocarcinomas a nivel de la ileostomía incluso años después de la coloproctectomía (un promedio de 20 años posteriores) y en el reservorio íleo pélvico, donde se han reportado casos de adenocarcinoma en el manguito residual y, aunque menos frecuentemente, en la bolsa ileal, razón por la cual debe realizarse seguimiento endoscópico de los mismos en forma anual o bienal según los hallazgos.
A nivel gástrico, los pólipos suelen ser hiperplásicos, del tipo glandulares fúndicos. Menos frecuentemente pueden encontrase pólipos adenomatosos, los cuales se ubican en el antro gástrico y deben manejarse endoscópicamente, si fuera posible; en caso contrario, y en presencia de displasia de alto grado o cáncer invasor, la gastrectomía se encuentra indicada.26

Tumores desmoides:12,27-30 son lesiones histológicamente benignas, que por su comportamiento expansivo y localización pueden comprometer diferentes órganos, provocando una oclusión intestinal, compresión ureteral y/o vascular que pueden ser graves.
Son infrecuentes en la población general, pero en las PAF se encuentran entre el 3,5 y el 59% de los pacientes, siendo, junto al cáncer periampular, la segunda causa de muerte en estos pacientes.
Los más frecuentes son los desmoides, que se presentan a nivel de la pared abdominal luego de la cirugía. También pueden presentarse como placas blanquecinas y fibróticas ubicadas generalmente en la superficie del mesenterio del intestino delgado, que pueden distorsionar la estructura del mismo, o como masas con distintos patrones de crecimiento que pueden ocupar toda la cavidad abdominal e incluso atravesar la pared abdominal.
Según la localización se clasifican en: extraabdominales; de la pared abdominal; intraabdominales; y transabdominales.
Los tumores desmoides son altamente recidivantes, y si bien existen varias líneas de tratamiento, ninguno ha demostrado altas tasas de curación.
Church y colaboradores,12 en el año 2005, propusieron una clasificación de los tumores desmoides que permite orientar el tratamiento, tomando en cuenta el tamaño, patrón de crecimiento y presencia de síntomas (tabla 31).
Los mejores resultados con cirugía se obtienen con la resección de los tumores desmoides ubicados en la pared abdominal. Para las lesiones ubicadas en el mesenterio, que involucran usualmente la raíz vascular y suelen presentar una forma difusa, la quimioterapia con adriamicina y viblastina ha sido una de las terapias más exitosas, aunque en la actualidad una alternativa es el uso de Imatinib. En estas lesiones intraabdominales complejas, que involucran los ejes vasculares y son difusas, se deben extremar los cuidados, tratando de evitar las resecciones.
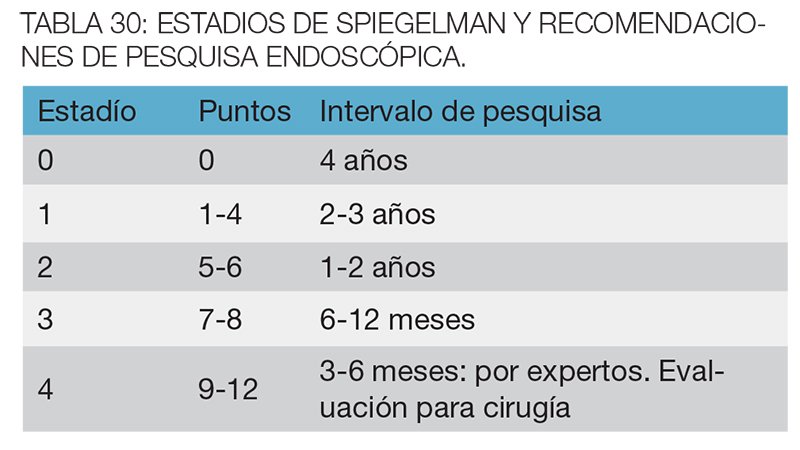
No existen aún recomendaciones de seguimiento, pero en aquellos individuos con PAF pertenecientes a familias con historia de desmoides, se encuentra indicada la tomografía de abdomen y pelvis preoperatoria.
En todos los pacientes con PAF el examen físico en busca de esta patología es de rutina. Ante su sospecha se deben realizar estudios de imágenes periódicos.
Tumores de tiroides:31-33 la incidencia de carcinoma no medular de tiroides en pacientes con PAF es del 1 al 12%, siendo más frecuente en mujeres (10-20 a 1).
La variante más común es el cáncer papilar con un patrón particular, denominado morular cribiforme, que es multicéntrico y bilateral en la PAF, por lo cual el tratamiento es la tiroidectomía total con un pronóstico excelente, con tasas de sobrevida del 95% a los 10 años.
El control sugerido por los distintos grupos es el examen físico anual de tiroides asociado a ecografía. En aquellos pacientes en los que se encuentren nódulos, está indicada la punción con aguja fina.31,34-35
Hepatoblastomas: se observa principalmente en niños menores de 5 años (aunque puede aparecer hasta los 16 años) con una incidencia de alrededor del 1%. En casos de familias con historia de hepatoblastoma, se debe realizar dosaje de α feto proteína cada 3 a 6 meses, examen físico y ecografía hepática anual desde los 6 meses hasta los 5-6 años. Si no hay antecedentes, su pesquisa aún se encuentra en debate.
Otras manifestaciones: tumores del SNC, lipomas, quistes sebáceos, quistes epidermoides, osteomas, dientes supernumerarios, engrosamiento de la cortical de huesos largos, hipertrofia del epitelio pigmentario de retina, se encuentran en un 60% de los pacientes con PAF. Fueron utilizados anteriormente como marcadores clínicos de la enfermedad, actualmente en desuso.
Diagnóstico de PAF
El diagnóstico de PAF se basa en una fuerte sospecha clínico-endoscópica (cientos a miles de pólipos colorrectales) y la confirmación anatomopatológica de su naturaleza adenomatosa. La confirmación de la presencia del gen responsable (APC, MUTHY, Pole, POLD2), si bien no es indispensable para el manejo de estos pacientes, sí resulta importante para el seguimiento familiar, sobre todo en la MAP por su patrón de herencia autosómica recesiva.
Si un paciente se presenta con síntomas y mediante la colonoscopia encontramos la PAF, se recomienda el examen físico en busca de manifestaciones extracolónicas, examen proctológico para evaluar el recto y decidir el tipo de cirugía, endoscopía digestiva alta de visión frontal y lateral (20-25 años) y ecografía de tiroides. Además, debe confeccionarse el árbol familiar (familigrama) y proceder a la citación de todos los familiares en riesgo para registrarlos, solicitarles rectosigmoideoscopía flexible o rígida en caso de formas floridas (afectación rectal segura) o colonoscopía completa en la forma atenuada.7,9,14,36-37
El estudio genético se solicita siempre en el contexto de un Registro o Unidad de Asesoramiento genético en oncología.9,34,36-41
Al paciente con diagnóstico clínico de PAF se le solicita la secuenciación completa del gen APC con informe de la mutación y de las variantes genéticas, o un panel de poliposis (secuenciación del gen APC y la búsqueda de las mutaciones en el gen MUTHY). En más del 80% de los casos, se halla el gen responsable.
Una vez detectada la mutación responsable de la enfermedad en esa familia, se puede realizar el análisis dirigido a buscar la mutación en todos los familiares en riesgos mayores de 10 años. Si no se encuentra, significa que no la heredaron y por lo tanto son individuos sanos con el mismo riesgo de CCR que la población general. En cambio, si heredaron la mutación familiar, los familiares en riesgo deben comenzar con colonoscopías periódicas y se debe indicar la cirugía ante la aparción de los pólipos.

Si no se puede realizar el estudio genético, se indica la pesquisa endoscópica a partir de los 10-12 años con rectosigmoideoscopía flexible o rígida en caso de formas floridas o colonoscopía en la forma atenuada (tabla 32).
Tratamiento de la PAF
El tratamiento quirúrgico en la PAF está indicado una vez diagnosticada la enfermedad para evitar el desarrollo de cáncer colorrectal. En los niños que presentan escasos pólipos, menores a 5 mm, puede contemporizarse posponiendo la cirugía hasta los períodos de recesos escolares o vacaciones. El momento correcto de la cirugía dependerá de la existencia de síntomas (2/3 de los pacientes con síntomas presentan CCR) que aceleren la indicación, del número y tamaño de los adenomas (a mayor número y mayor tamaño, mayor riesgo de malignizacion), de la presencia de displasia de alto grado o cáncer y en pacientes con mutaciones relacionadas a mayor riesgo de CCR. Todas estas son razones para que una vez realizado el diagnóstico el paciente sea operado a la brevedad.2,7,34
Si bien existen diversas opciones quirúrgicas, cada una de ellas presenta indicaciones precisas. En la actualidad, el abordaje laparoscópico es la opción más utilizada (tabla 33).42-46
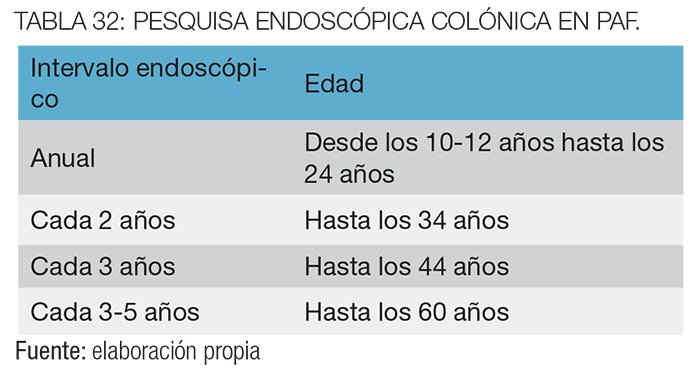

No se recomienda realizar este procedimiento en pacientes con mala adherencia al seguimiento médico, ya que el riesgo de cáncer del recto remanente en este grupo es alto (12-29%).
-
Coloproctectomía total con reservorio ileal y anastomosis ileoanal.
Este procedimiento está indicado en pacientes con severa afectación colónica y rectal (más de 20 pólipos rectales), cáncer de recto curable que no invada esfínteres (estadios tempranos), recto con pólipos mayores a 3 cm o con presencia de displasia severa, preferencia del paciente y ante la necesidad de proctectomía luego de una anastomosis ileorrectal previa. Algunos autores sugieren que en aquellos individuos con mutaciones entre los codones 1.250 y 1.464, asociadas a mayor riesgo de cáncer de recto, esta técnica sería de elección aunque el recto inicialmente no se encuentre muy afectado.
El procedimiento consiste en la resección del colon y recto y la confección de un reservorio con el intestino delgado (bolsa o pouch ileal generalmente en J) con anastomosis ileoanal.
En mujeres jóvenes en edad fértil que deseen ser madres, se recomienda retardar lo más posible la coloproctectomía (siempre que no exista neoplasia o displasia de alto grado rectal), debido a que la disección pélvica ocasiona una disminución de la fertilidad.
Como se describió en los párrafos previos, luego de la cirugía debe continuarse con controles endoscópicos.7, 34,47-48
La presencia de desmoides intraabdominales puede hacer peligrar la conversión de una ileorrectoanastomosis a un reservorio ileal. Por ello algunos autores sugieren que en casos con historia familiar de desmoides, la elección también sería una coloproctectomía con reservorio ileal aun con recto poco afectado. -
Colectomía total con ileorrectoanastomosis (IRA)
La colectomía total con ileorrectoanastomosis se encuentra indicada en pacientes que presentan un recto con escasa afectación (20 pólipos) –evaluado por un coloproctólogo experto en PAF– y en pacientes con un cáncer de colon avanzado.34,47
Al conservarse el recto, el paciente debe controlarse con endoscopía periódicamente y se deben resecar todos los pólipos mayores a 5 mm hallados en estos estudios y enviarse a anatomía patológica.7 Cuando la enfermedad rectal es severa, o aparezcan lesiones que no puedan ser resecadas endoscópicamente, se debe proceder a la proctectomía con o sin restauración del tránsito mediante un reservorio ileal o ileostomía, según sea posible. -
Coloproctectomía con ileostomía definitiva a lo Brooke
Se indica en aquellos pacientes con cáncer rectal bajo que invade esfínteres, estadios avanzados de cáncer de recto, personas con afección de la continencia anal y en pacientes con indicación de coloproctectomía e imposibilidades técnicas de realizar un reservorio ileal (obesos, presencia de desmoides de mesenterio).47
Estos individuos deben continuar con controles cada dos años de la ileostomía, ya que existe riesgo de desarrollar adenocarcinoma a nivel del ostoma.
La poliposis atenuada (PAFA) es una variante fenotípica de la PAF producida generalmente por mutaciones en los extremos 5´ y 3´ del gen APC. Se caracteriza por la aparición de los pólipos y el cáncer colorrectal generalmente en los 10 años posteriores al promedio de aparición en las formas clásicas y por presentar una mayor afectación del colon proximal. Esta es la razón por la cual en este grupo se indica la colonoscopía para la pesquisa, a partir de los 20 años, cada 2 años.14, 34 El riesgo de desarrollar CCR alcanza al 80% a lo largo de la vida y presenta un riesgo similar de desarrollar manifestaciones extracolónicas que la PAF clásica.
En este grupo, la endoscopia terapéutica puede tener un lugar para el tratamiento de los casos con escasa afectación colorrectal, reservándose la cirugía para aquellos en los cuales el número de pólipos impide su competa resección, son de gran tamaño o presentan displasia de alto grado o neoplasia.
En cuanto a las manifestaciones extracolónicas, se deben seguir las mismas recomendaciones que en las formas clásicas.34
Poliposis asociada a MUTYH
Esta forma de poliposis presenta un patrón de herencia autosómica recesiva (mutaciones bialélicas homocigotas o heterocigotas compuestas). Fenotípicamente, se puede expresar como una poliposis atenuada o clásica con 10 o hasta unos pocos cientos de adenomas y excepcionalmente con más de 500 pólipos (formas floridas).10,49-53 Debido a que pueden coexistir pólipos serratos, también se puede expresar como una poliposis serrata.1,34,39
Se produce por mutaciones bialélicas en el gen MYH, siendo las dos más comúnmente halladas Y179C y G396D (antes denominadas Y165C y G382D).
El riesgo para CCR y la forma de presentación es similar a las formas atenuadas, con mayor afectación del colon proximal y una edad de aparición de alrededor de los 45 años.
Se ha descripto la presencia de manifestaciones extra intestinales, tales como adenomas duodenales, pólipos glandulares fúndicos gástricos y quistes epidermoideos, aunque con menor frecuencia que en la PAF. También se ha descripto un riesgo incrementado para otros tumores extra intestinales, tales como los cánceres de ovario, vejiga, piel y mama.
El seguimiento y tratamiento de los pacientes con mutaciones bialélicas es similar a la PAFA.1,7,34
Las mutaciones monoalélicas MUTYH se encuentran en el 1 al 2% de la población general y se asocian a un riesgo incrementado de CCR aún incierto (algunos estudios estiman que este riesgo es de 1.5 a 2 sobre la población general).10,39,54
No existe consenso sobre el seguimiento de los portadores monoalélicos, pero algunos autores sugieren realizar una colonoscopía a los familiares de primer grado 10 años antes del caso de CCR mas joven y luego cada 5 años.10
Poliposis asociada a corrección de lectura de polimerasa (PPAP) o poliposis asociadas a POLE o POLD110,55-57
Esta poliposis se presenta con adenomas colorrectales en número variable de 10 a 100, con un mayor riesgo de CCR a edades más tempranas. Se transmite en forma autosómica dominante, con una penetración que parecería ser alta y se asocia a mutaciones en los genes POLE y POLD 2.
Aún no existen recomendaciones sobre el tratamiento de la PPAP, ya que la frecuencia de los pólipos, del cáncer y de las manifestaciones extracolónicas aún no ha sido bien determinada, pero dependiendo del número de pólipos, el tratamiento endoscópico o la colectomía parecerían ser razonables.
En cuanto al seguimiento de las manifestaciones extracolónicas, en las mujeres portadoras del gen mutado POLD1 se debería contemplar la realización de una ecografía pélvica y biopsia selectiva de endometrio, ya que esta mutación se asocia a mayor riesgo de cáncer de endometrio.
Poliposis adenomatosa asociada a mutaciones bialélicas de los MMR (genes reparadores de daños del ADN) o déficit constitucional de genes reparadores de daños (CMMRD)5,10,55
Esta poliposis se produce por mutaciones en los genes reparadores de daño del ADN (más frecuentemente PMS2 y MSH6) y su herencia es autosómica recesiva. Se presenta como una oligo poliposis con un número variable de adenomas (entre 10 y 100), con manchas café con leche en la piel y puede asociarse a tumores del tracto digestivo (colon 40%, intestino delgado 12%), de cerebro a enfermedades oncohematológicas (principalmente linfoma no Hodgkin), cáncer de endometrio y del tracto urinario.
El seguimiento de los portadores de mutaciones bialélicas en los MMR debe realizarse con:
- Colonoscopia cada 1 -2 años desde los 8 años de edad.
- Endoscopia digestiva alta y video cápsula endoscópica anual a partir de los 10 años.
Síndrome de Poliposis Juvenil (PJ)
La Poliposis Juvenil se produce por mutaciones germinales en alguno de los siguientes genes: SMAD4 (cromosoma 18q1.1); BMPR1A (cromosoma 10q22-23); ENG (cromosoma 9q 4.1). Los dos primeros son los más frecuentemente mutados. Su herencia es autosómica dominante y se caracteriza por la presencia de pólipos hamartomatosos del tipo juveniles en el tracto gastrointestinal, generalmente de aparición temprana (en la primera o segunda década de la vida). Su incidencia se encuentra entre 1:100.000 a 1:160.000 individuos.10,56,58-63
Al igual que los otros síndromes de poliposis, esta enfermedad se asocia a un riesgo incrementado de desarrollar tanto cáncer colorrectal (9 al 70%) como tumores malignos de otros órganos –estómago (21%), páncreas, duodeno e intestino delgado.
Se ha descripto que las mutaciones en el gen SMAD4 se asocian a telangiectasia hereditaria hemorrágica (THH) (Síndrome de Osler Weber Rendú). Por lo tanto en aquellas familias portadoras de mutaciones en este gen debe realizarse el estudio genético a sus hijos dentro de los primeros 6 meses de vida, debido al riesgo de THH.10,58-63
Criterios diagnósticos del Síndrome de Poliposis Juvenil64
- Al menos 3 a 5 pólipos juveniles rectocolónicos; o
- múltiples pólipos juveniles en el tracto gastrointestinal; o
- cualquier número de pólipos juveniles y una historia familiar de poliposis juvenil.
Pesquisa y vigilancia en Poliposis Juvenil58-62
La pesquisa es recomendada para todos los individuos con sospecha clínica o confirmada por diagnóstico genético de todos los familiares en riesgo (tabla 34). En caso de contar con la determinación genética, esta debe realizarse en una primera instancia al paciente con diagnóstico clínico (caso índice), y en caso de hallarse la mutación, realizar la misma a los familiares en riesgo.
Debe remarcarse que el estudio genético no es indispensable para el seguimiento de estas familias. Si no se pudiera realizar, ya sea por el costo, porque no hay algún familiar afectado vivo o porque en la familia no se pudo hallar la mutación, debe practicarse la pesquisa endoscópica, mediante endoscopías digestivas altas y bajas a partir de los 10-15 años.
El tratamiento de los individuos afectados dependerá de la posibilidad del control endoscópico del colon. Si no es factible por el número o el tamaño de los pólipos, la presencia de cambios adenomatosos o de displasia de alto grado o en individuos con poliposis colónica sintomática, la cirugía se encuentra indicada con las mismas variantes que en la PAF dependiendo del grado de afectación rectal: colectomía total con ileorrectoanastomosis o coloproctectomía con reservorio ileal al igual que en la PAF.
Manifestaciones extracolónicas en la Poliposis Juvenil. Vigilancia
Tracto digestivo alto: los pólipos juveniles se encuentran en un 14% en el estómago, 7% en yeyuno íleon y 7% en duodeno y están asociados también un mayor riesgo de desarrollar cáncer gástrico. La pesquisa se realiza mediante endoscopías digestivas altas desde los 10 a 15 años de edad, con una frecuencia anual si hay pólipos, o cada 2 a 3 años si no los hubiera. En aquellos pacientes que presentan displasia de alto grado, cáncer gástrico o una poliposis gástrica severa, que no pueda ser tratada endoscópicamente, debe contemplarse la gastrectomía total o subtotal.
En caso de severa afectación duodenal, existencia de anemia inexplicable, enteropatía perdedora de proteínas u otros síntomas atribuibles a la afectación de intestino delgado, se encuentra indicado su estudio con enteroscopia, cápsula endoscópica, entero tomografía o entero resonancia.
Malformaciones vasculares: por el riesgo de desarrollar malformaciones vasculares asociadas a THH, se deben comenzar los controles dentro de los primeros 6 meses de vida con el examen cardiovascular, el hemograma y la evaluación de síntomas abdominales.
Síndrome de Peutz Jeghers
El Síndrome de Peutz Jeghers es una poliposis hamartomatosa gastrointestinal hereditaria, que se produce por mutaciones germinales en el brazo corto del cromosoma 19 – 19p13.3, en el gen LKB1 – STK11 con herencia autosómico dominante. Se presenta en 1:50.000 a 1:200.000 nacidos vivos.10,56,58-60
Este síndrome se caracteriza por la asociación de pólipos hamartomatosos asociados a hiperpigmentación de la mucosa oral y de la piel de las palmas de las manos y plantas de los pies.58-60
Los pólipos son más frecuentes en el intestino delgado (96%), seguidos de colon (27%), recto (24%) y estómago (24%), mientras que los sitios de malignizacion, en orden de frecuencia, son: colon y el recto (39%), estómago (29%), intestino delgado (13%), páncreas (11%). Entre los tumores extraintestinales se ha descripto: cáncer de pulmón (7-17%), el cáncer de mama (24-54%), el tumor de ovario de la cuerda sexual con túbulos anulares (SCTAT) (18-21%), el adenoma maligno de útero (adenocarcinoma bien diferenciado de endocervix: 10-23 %), tumor mucinoso de ovario y el tumor de células de Sertoli.10,58-60
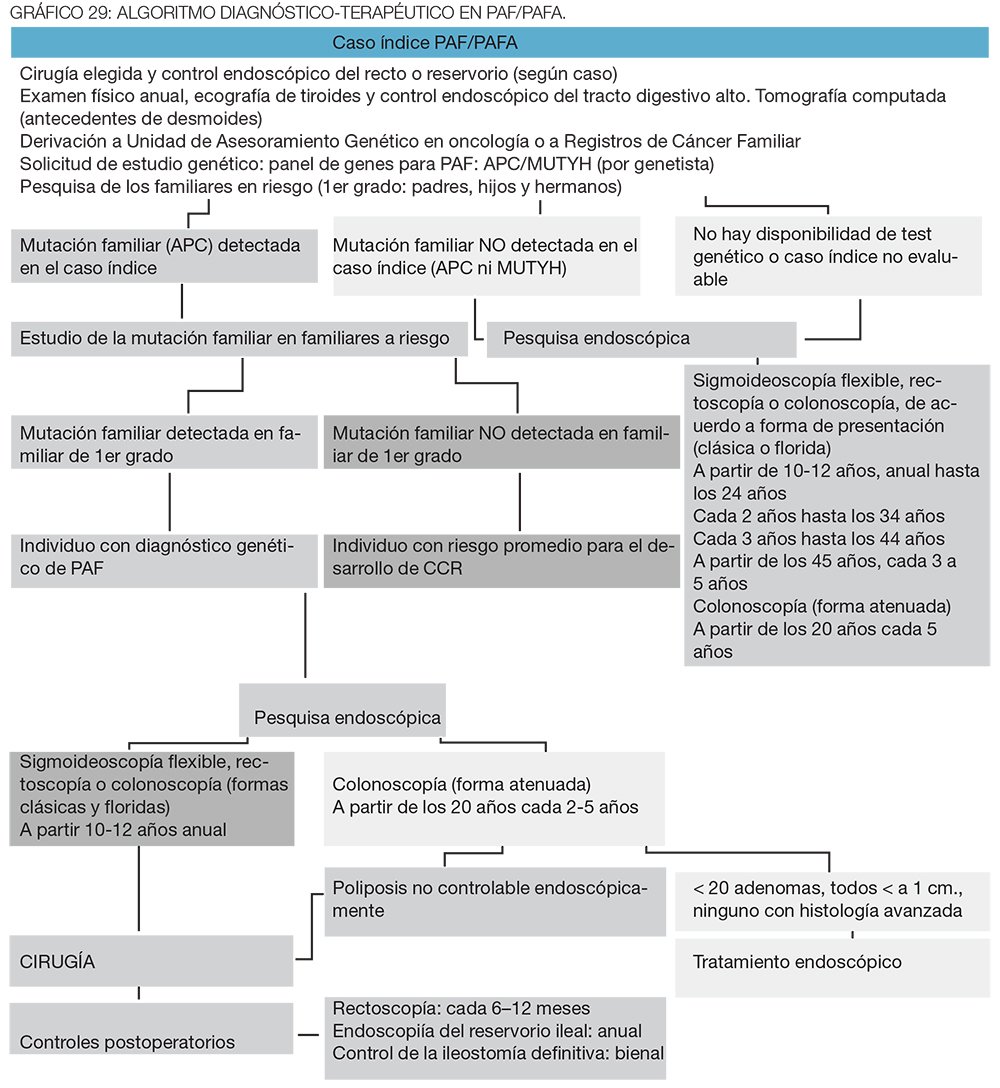
Su diagnóstico por sospecha clínica es alto debido a que estos pacientes concurren con una historia previa de cuadros de oclusión o suboclusión intestinal que en la mayoría de los casos requirieron un abordaje quirúrgico; además, al examen físico se encuentran las manchas melánicas descriptas.
El diagnóstico clínico de Síndrome de Peutz Jeghers puede ser realizado si un individuo cumple dos o más de los siguientes criterios:64
- Dos o más pólipos hamartomatosos de tipo Peutz Jeghers (confirmados histológicamente) en el intestino delgado.
- Hiperpigmentación mucocutánea en boca, labios, nariz, ojos, genitales o dedos.
- Historia familiar de Síndrome de Peutz Jeghers.
Pesquisa y vigilancia en Síndrome de Peutz Jeghers10,58-60
La pesquisa es recomendada para los individuos con sospecha clínica o confirmación diagnóstica por estudio genético y para todos los familiares en riesgo. En caso de contar con la determinación genética, esta debe realizarse en primera instancia al paciente con diagnóstico clínico de la enfermedad, y, en caso de hallarse la mutación, recién allí se ofrece realizarla a los familiares en riesgo.

Debemos remarcar que el estudio genético no es indispensable para el seguimiento de estas familias. Si este no se puede realizar por el costo, porque no hay algún familiar afectado vivo o porque en la familia no se pudo hallar la mutación, debe realizarse la pesquisa endoscópica, mediante endoscopías digestivas altas y bajas a partir de los 8 años (algunos autores sugieren a partir de la adolescencia).
Si se encuentran pólipos, deben resecarse y repetirse la endoscopia cada 2-3 años hasta los 50 años. Si no se detectan pólipos, se repite a los 18 años de edad, o antes en caso de aparición de síntomas. A partir de los 50 años de edad, repetir cada 1 o 2 años, por el aumento del riesgo de CCR a partir de esta edad.
Si por el número o el tamaño las lesiones no pudieran resecarse, si existieran cambios adenomatosos o displasia de alto grado o una poliposis colónica sintomática, la colectomía total con ileorrectoanastomosis se encuentra indicada.
Manifestaciones extracolónicas en Síndrome de Peutz Jeghers. Vigilancia (tabla 35)
Tracto digestivo alto: el tracto digestivo alto debe comenzar vigilarse con endoscopías digestivas altas y el intestino delgado mediante cápsula endoscópica, tránsito de intestino delgado, entero tomografía o entero resonancia, según la disponibilidad. Debe repetirse de acuerdo a los hallazgos, si no cada 2-3 años o antes si aparecen síntomas. Si no hay pólipos en el estudio inicial, el examen debe repetirse a los 18 años o antes si aparecen síntomas, repitiéndolo cada 3 años. En aquellos pacientes que presenten pólipos mayores de 1 cm o sintomáticos, si es posible, deben resecarse por endoscopía; cuando no sea viable o existan pólipos grandes en intestino delgado, la cirugía con enterotomía y polipectomías múltiples puede estar indicada (con enteroscopía intraoperatoria).
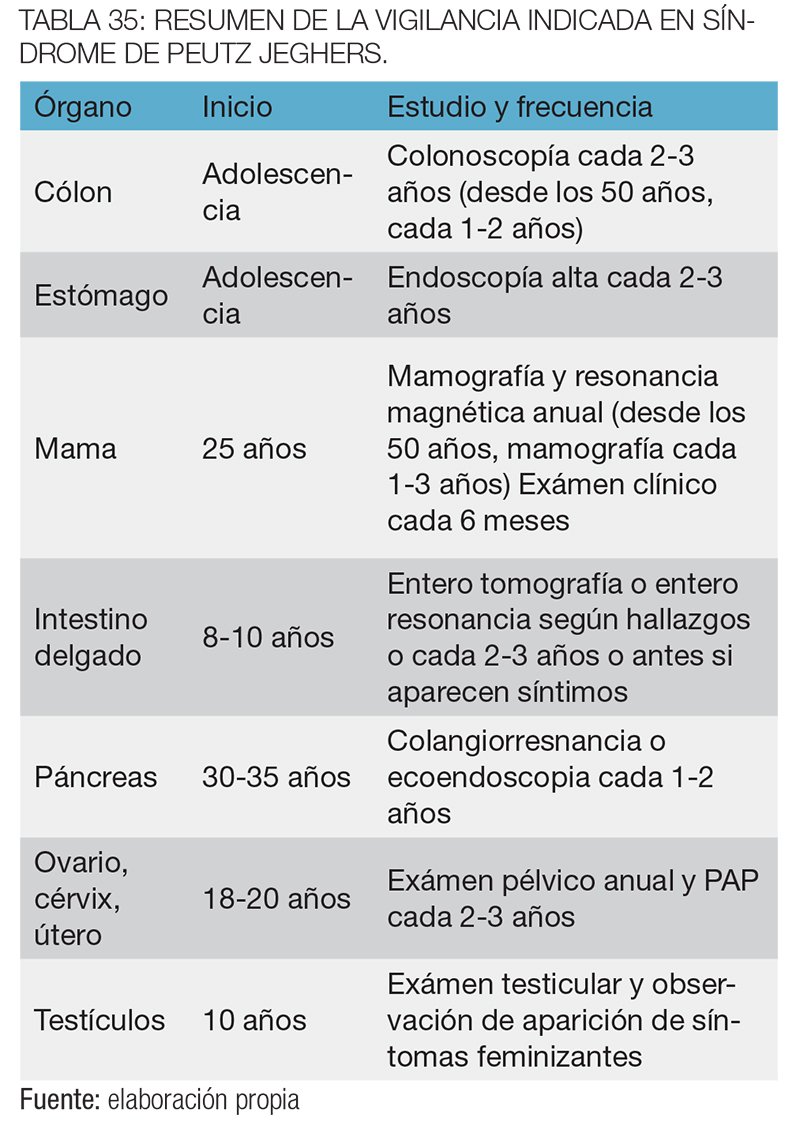
Otras manifestaciones: deben realizarse controles hematológicos (hemograma y análisis de función hepática anual), examen clínico anual, colangiorresonancia o eco endoscopía cada 1-2 años, a partir de los 30-35 años (pesquisa de cáncer de páncreas). En varones, examen testicular anual desde los 10 años de edad (si hubiera algún hallazgo sospechoso ultrasonografía testicular); en mujeres, realizar examen pélvico anual y PAP cada 2-3 años (pesquisa para cáncer de cuello uterino) a partir de los 18-20 años de edad y mamografía o resonancia magnética de mama anual entre los 25 y 50 años y mamografía desde los 50 años cada 1 a 3 años.

Los familiares de primer grado de pacientes con Síndrome de Peutz Jeghers deben ser evaluados anualmente desde su nacimiento con examen físico, objetivar la presencia de manchas melánicas o de pubertad precoz y en los varones buscar tumores testiculares.
Síndrome de poliposis mixta hereditario65-68
Es un raro síndrome autosómico dominante caracterizado por el desarrollo de un número variable de pólipos de distinta histología, incluyendo adenomas, pólipos hiperplásicos o serratos, pólipos juveniles y pólipos mixtos a nivel colónico. En algunos casos su diagnóstico diferencial con un síndrome de poliposis juvenil puede ser indistinguible, pudiendo también confundirse con una poliposis hiperplásica. La edad media de aparición de los pólipos es alrededor de los 28 años.
Aún no se ha reportado la asociación con manifestaciones extracolónicas y el riesgo exacto de desarrollo de cáncer colorrectal se desconoce, aunque estaría aumentado. Se produce por mutaciones en el gen GREM 1, y el tratamiento es similar a la PAF, dependiendo del número tamaño e histología de los pólipos.
Síndromes de Tumores Hamartomatosos asociados a mutaciones en el gen PTEN
Síndrome de Cowden10,56,58-59 síndrome de transmisión autosómica dominante caracterizado por múltiples lesiones hamartomatosas y neoplasias de origen endodérmico, mesodérmico y ectodérmico que afectan a diversos sistemas y órganos.

Lesiones mucocutáneas: especialmente en piel (pápulas faciales, queratosis sacra, fibromas escleróticos múltiples) y membranas mucosas (papilomatosis de la mucosa oral).
Anormalidades tiroideas: localización extra cutánea más frecuente, siendo el bocio y los adenomas las lesiones más comunes. El adenocarcinoma folicular de tiroides ha sido reportado en el 3 al 12% de los pacientes.
Anormalidades mamarias: el carcinoma de mama es el tumor maligno más frecuente. Es usualmente bilateral y de tipo ductal.
Pólipos gastrointestinales: ocurren en el 40 al 70% de los pacientes. Pueden incluir manifestaciones a nivel del sistema nervioso central como macrocefalia, gangliocitoma de cerebelo y algunas veces retardo mental.
Síndrome de Bannayan-Riley-Ruvalcaba (SBRR):58-59
Síndrome congénito que se caracteriza por macrocefalia, disfunción cognitiva y motora, lipomas viscerales y subcutáneos, hemangiomas, manchas pigmentarias peneanas y pólipos de tipo juvenil en el colon. No ha sido documentado aún un incremento del riesgo para CCR (tablas 36 y 37).
Tanto el Síndrome de Cowden como el SBRR presentan una alteración en el brazo largo del cromosoma 10 (10q21-23) en el gen PTEN.
Síndrome de Poliposis Serrata
La Poliposis Hiperplásica/Serrata es un raro síndrome cuya incidencia es de alrededor de 1:100.000. Presenta un riesgo incrementado para el desarrollo de CCR, que varía de 0 a 50%.69-77 En series pequeñas, se ha reportado una frecuencia de CCR mayor al 69%, mientras que en las grandes series fue del 37%. Si bien el riesgo es mayor, continúa siendo indefinido. Además, parecería existir un riesgo incrementado para el desarrollo de CCR en los familiares de primer grado de pacientes con diagnóstico de Poliposis Serrata.10,69,78 Boparai y colaboradores79 han encontrado un incremento del riesgo relativo de 5.4 en los familiares de primer grado.
Se ha descripto una importante relación entre el hábito tabáquico y el desarrollo de Síndrome de Poliposis Hiperplásica.
Su diagnóstico es clínico y debe ser considerado cuando un individuo presenta al menos uno de los siguientes criterios empíricos propuestos por la Organización Mundial de la Salud (OMS):64,80-84
- Al menos 5 pólipos serratos proximales al colon sigmoideo, con dos o más de ellos mayores a 1 cm.
- Cualquier número de pólipos serratos proximales al colon sigmoideo en individuos que tienen un familiar de primer grado afectado de Poliposis Serrata.
- Más de 20 pólipos serratos de cualquier tamaño, pero distribuidos en todo el colon.
Nomenclatura: dentro de la Poliposis Serrata se incluirán, para el conteo de las lesiones, pólipos hiperplásicos, adenomas/pólipos serratos sésiles, adenomas serratos tradicionales.
Se describen dos variantes clínicas:
- Tipo 1: poliposis adenomatosa serrata con la presencia de distintos tipos de pólipos hiperplásicos (lesiones serratas sésiles, adenoma serrato tradicional, pólipos mixtos o lesión serrata con displasia, pólipos hiperplásicos y adenomas convencionales). Esta se asocia a importante riesgo de CCR.
- Tipo 2: poliposis hiperplásica/serrata, la cual comprende pequeños pólipos hiperplásicos clásicos (menores a 5 mm), con bajo riesgo de malignidad.
Síndrome de Jass: variante de Poliposis Serrata familiar, descripta en el año 2005. Son familias con predisposición hereditaria a desarrollar CCR por la vía serrata. Puede presentar inestabilidad en microsatélites (IMS) y frecuentemente se asocia a mutaciones somáticas del gen BRAF en el tumor y presencia de pólipos adenomatosos y serratos. Se ha ligado este síndrome a mutaciones en el cromosoma 2 (2q32.2-q33.3).83
Vigilancia en Poliposis Serrata10,80-84
Una vez identificado el paciente, se debe indicar la colonoscopía y la evaluación de la familia, considerando en riesgo a los familiares de primer grado, quienes deberán comenzar la pesquisa a los 40 años de edad o 10 años antes de la edad del caso índice (en el caso con CCR al momento del diagnóstico) o a la edad del caso más joven diagnosticado (si dicho caso no presentará CCR). Los padres o hermanos mayores deberán realizarse la colonoscopía cuando se hace el diagnóstico en el caso índice.
La vigilancia de los individuos será cada 1-3 años con resección de todos los pólipos mayores a 0.5 cm.
En los familiares de primer grado, se indica colonoscopía cada 5 años si no se encontraran lesiones polipoideas en el estudio de base, y de preferencia con técnicas endoscópicas de magnificación.
En aquellos pacientes que al momento del diagnóstico presenten un CCR asociado, se los debe estadificar de la misma manera que se realiza con el cáncer esporádico.
No requiere otro tipo de estudios complementarios.
El estudio genético de los pacientes con Poliposis Serrata no está indicado como de rutina, pero debe evaluarse la posibilidad de realizar el estudio de mutaciones en el gen MUTYH en aquellos pacientes en que exista la presencia de adenomas asociados a las lesiones aserradas.
En esta entidad las recomendaciones se basan en opiniones de expertos.
Cirugía en la Poliposis Serrata
En este grupo, el tratamiento es la resección endoscópica de todos los pólipos mayores o iguales a 5 mm, con un intervalo dependiente del número y tamaño de los pólipos (1 a 3 años). Se debe considerar la colectomía si el tratamiento o el seguimiento endoscópico son inadecuados, o si existe displasia de alto grado. Aunque se sugiere considerar la colectomía total, aún no existe un fuerte consenso para elegir entre esta y una resección segmentaria.
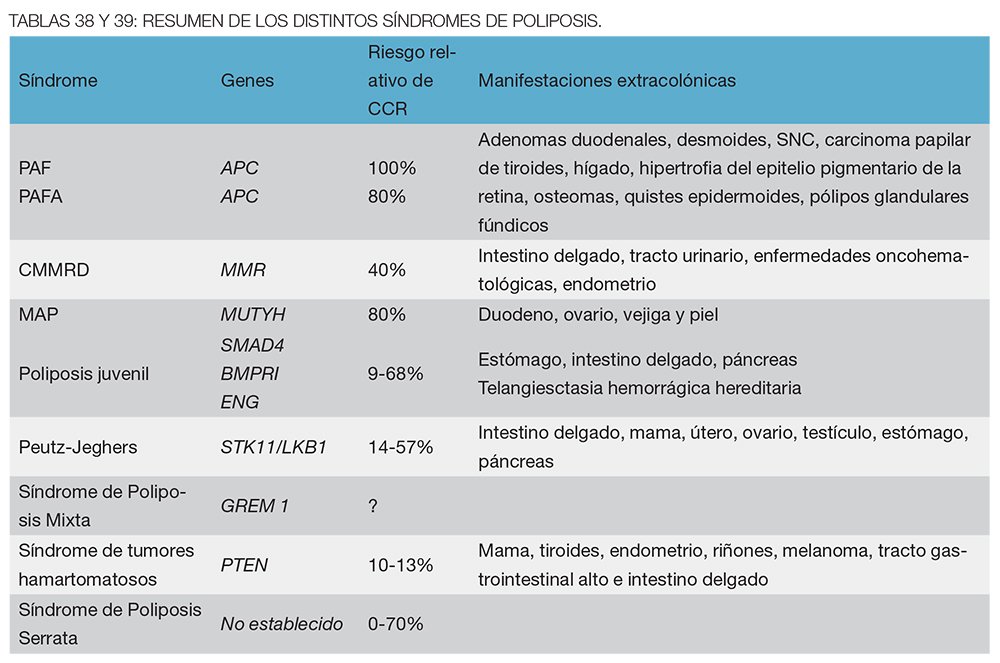

BIBLIOGRAFÍA
- Aretz, S. (2010): “The Differential Diagnosis and Surveillance of Hereditary Gastrointestinal Polyposis Syndromes”. Em: Dtsch Arztebl Int, 107(10): 163-173.
- Al Sukhni, W.; Aronson, M. y Galliger, S. (2008): “Hereditary colorectal cancer syndromes: Familial adenomatous polyposis and Lynch syndrome”. En: Surg Clin N Am, 88: 819-844.
- Bullow, S. (2003): “Results of national registration of familial adenomatous polyposis”. En: Gut, 52: 742-746.
- Gryfe, R. (2009): “Inherited Colorectal Cancer Syndromes”. En: Clinics in colon and rectal surgery, 22(4): 198-208.
- Vasen, H.F.A.; Tomlinson, I. y Castells, A. (2015): “Clinical management of hereditary colorectal cáncer”. En: Nat Rev Gastroenterol Hepatol, 12: 88-97.
- Ellis, C.N. (2008): “Colonic adenomatous polyposis syndromes: Clinical management”. En: Clin Colon Rectal Surg, 21: 256-262.
- Cairs, S.R.; Scholefield, J.H.; Steele, R.J. et al. (2010): “Guidelines for colorectal cancer screening and surveillance in moderate and high risk groups (update from 2002)”. En: Gut, 59: 666-689.
- Jewel Samadder, N. y Jasperson, K. (2015): “Hereditary and common familial colorectal cancer: evidence for colorectal screening”. En: Dig Dis SCI, 60: 737-747.
- Half, E.; Bercovich, D. y Rozen, P. (2009): “Familial Adenomatous polyposis”. En: Orp J Rare Diseases, 4: 1-23.
- Syngal, S.; Brand, R.E.; Church, J.M. et al. (2015): “ACG Clinical Guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes”. En: Am J Gastroenterol, 110: 223-262.
- Jung, I.; Gurzu, S. y Turdean, G.S. (2015): “Current status of familial gastrointestinal polyposis syndromes”. En: World J Gastrointest Oncol, 7(11): 347-355.
- Church, J.; Lynch, C.; Neary, P. et al. (2005): “A desmoid Tumor-Staging System Separetes Patients with Intra-Abdominal, Familial adenomatous Polyposis-Associated Desmoid Disease by Behavior and Prognosis”. En: Dis Colon Rectum, 51: 897-901.
- Valle, L.; Hernandez Illan, E.; Bellido, F. et al. (2014): “New insights into Pole and PolD1 germline mutations in familial colorectal cancer and polyposis”. En: Hum Molecular Genetics, 23(13): 3506-3512.
- Vasen, H.F.A.; Moslein, G.; Alonso, A. et al. (2008): “Guidelines for the clinical management of familial adenomatous polyposis (FAP)”. En: Gut, 57: 704-713.
- Garzon Benavides, M.; Pizarro Moreno, A.; García Lozano, R. et al. (2010): “Andalusian registry for familial adenomatous polyposis. Analysis of patients included”. En: Rev Esp Enferm Dig, 102(11): 653-657.
- Berk, T.; Cohen, Z. y Cullen, J.B. (1981): “Familial polyposis and the role of the preventive registry”. En: CMA, 124: 1427-1428.
- Reyes Moreno, J.; Vicens, D.G.; Vanrell, M. et al. (2007): “Mejoría de la supervivencia de la poliposis adenomatosa familiar después del establecimiento de un registro”. En: Med Clin, 129(2): 51-52.
- Barrow, P.; Khan, M.; Lallo, F. et al. (2013): “Systematic review of the impact of registration and screening on colorrectal incidente and mortality in familial adenomatous polyposis and Lynch syndrome”. En: Br J Surg, 100: 1719-1731.
- Mallinson, E.K.L.; Newton, K.F.; Bowen, J. et al. (2010): “The impact of screening and genetic registration on mortality and colorectal cancer incidence in familial adenomatous polyposis”. En: Gut, 59: 1378-1382.
- Dalpatadu, K.U.A.; Anwar, N.; Wijesuriya, S.R.E. et al. (2006): “Use of a familial adenomatous polyposis registry”. En: Ceylon Medical Journal, 56: 66-69.
- Gallagher, M.; Phillips, R.K.S. y Bullow, S. (2006): “Surveillance and management of upper gastrointestinal disease in Familial Adenomatous Polyposis”. En: Familial Cancer, 5: 263-273.
- Brosens, L.A.A.; Keller, J.J.; Offerhaus, G.J.A. et al. (2005): “Prevention and management of duodenal polyps in familial adenomatous polyposis”. En: Gut, 54: 1034-1043.
- Skipworth, J.R.; Morkane, C.; Raptis, D.A. et al. (1996): “Pancreaticoduodenectomy for advance duodenal and ampullary adenomatosis in familial adenomatous polyposis”. En: HPB, 13: 342-348
- Stern, H.S. (1996): “The Canadian familial adenomatous polyposis registry: past, present and future”. En: J R Soc Med, 89: 153-154.
- Delpero, J.R.; Turrini, O. y Ewald, J. (2014): “Duodenectomue totale sans pancreatectomie pour polypose adenomateuse familiale (avec video)”. En: Journal de Chirurgie Viscerale, 151: 487-488.
- Will, O.C.C.; Man, R.F.; Phillips, R.K.S. et al. (2008): “Familial adenomatous polyposis and the small bowel: a loco regional review and current management strategies”. En: Pathology Research and Practice, 204: 449-458.
- Wang, X.Y.; Wei, X.; Dai, Y.H. et al. (2013): “Large gastric folds arising in polyposis syndromes”. En: Rev Esp ENferm Dig, 105(7): 429-432.
- Escobar, C.; Munker, R.; Thomas, J.O. et al. (2012): “Update on desmoid tumors”. En: Ann of Oncol, 23: 562-569.
- Kasper, B.; Ströbel, P. y Hohenberger, P. (2011): “Desmoid Tumors: Clinical Features and Treatment Options for advanced Disease”. En: The Oncologist, 16: 682-693.
- Calvert, G.T.; Monument, M.J.; Burt, R.W. et al. (2012): “Extra abdominal desmoid tumors associated with familial adenomatous polyposis”. En: Sarcoma, 12: 1-11.
- Sloane, J.; Ranchod, P.; Willams, G. et al. (2013): “Familial adenomatous polyposis: not all mases are desmoids”. En: Familial Cancer, 12: 525-528.
- Herraiz, M.; Barbesino, G.; Faquin, W. et al. (2007): “Prevalence of thyroid Cancer in Familial Adenomatous Polyposis Syndrome and the Role of Screening Ultrasound Examinations”. En: Clin Gastroenterol Hepatol, 5: 367-373.
- Liyanapathirana, N.; Seneviratne, S.A.; Samarasekera, D.N. (2015): “A distinct variant of papillary thyroid carcinoma indicating familial adenomatous polyposis (FAP): a case report and brief review”. En: BMC Res Notes, 8: 795-798.
- Cetta, F. (2015): “FAP associated papillary thyroid carcinoma: a peculiar subtype of familial non medullary thyroid cáncer”. En: Pathology Research International, 1-6.
- Bonis, P.; Ahnen, D. y Axel, L. (2011): “Familial adenomatous polyposis and MYH associated polyposis: Screening and management of patients and families”. En: Acta Gastroentrolg Belg, 74(3): 421-426.
- Groen, E.J.; Roos, A.; Muntinghe, F. et al. (2008): “Extra intestinal manifestations of familial adenomatous polyposis”. En: Ann of Surg Oncol, 15(9): 2439-2450.
- Burt, R.W. ; Bathel, J.S.; Drini, M. et al. (2010): “Colorectal cancer screening. Clinical Practice Guidelines in Oncology”. En: JNCCN, 8(1): 8-61.
- Levin, B.; Lieberman, D.; Mc Farland, T. et al. (2000): “Colorectal carcinomas arising in the hyperplastic polyposis sybdrome progress through the chromosomal instability pathway”. En: AJP, 157: 385-392.
- Stoffel, E.M.; Mangu, P.B.; Gruber, S.B. et al. (2014): “Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice guideline endorsement of the familial risk colorectal cancer: European Society for Medical Oncology Clinical Practice Guidelines”. En: J Clin Oncol, 33: 209-217.
- Leoz, M.L.; Carballal, S.; Moreira, L. et al. (2015): “The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management”. En: The Application of Clinical Genetic, 8: 96-107.
- Lynch, P. (2013): “When and how to perform genetic testing for inherited colorectal cancer syndromes”. En: J Natl Compr Canc Netw, 11: 1577-1583.
- Fernández Suárez, A.; Cordero Fernández, C.; García Lozano, R. et al. (2005): “Clinical and ethical implications of genetic counseling in familial adenomatous polyposis”. En: Rev Esp Enferm Dig, 97(9): 654-665.
- Campos, F.G. (2014): “Surgical treatment of familial adenomatous polyposis: dilemmas and current recommendations”. En: World J Gastroenterol, 20(44): 16620-16629.
- Warrier, S.K.; Kalady, M.F. (2012): “Familial adenomatous polyposis: challenges and pitfalls of surgical treatment”. En: Clin Colon Rectan Surg, 25: 83-89.
- Tulchinsky, H.; Keidar, A.; Goldman, G. et al. (2005): “Surgical treatment and long term outcome of patient with familial adenomatous polyposis: 16 years experience at the Tel Aviv Sourasky Medical Center”. En: IMAJ, 7: 82-85.
- Kartheuser, A.; Stangherlin, P.; Brandt, D. et al. (2015): “Restorative proctocolectomy and ileal pouch anal anastomosis for familial adenomatous polyposis revisited”. En: Familial Cancer, 5: 241-260.
- Kalady, M. y Church, J. (2015): “Prophylactic Colectomy: rationale, indications and approach”. En: J Surg Oncology, 115: 112-117.
- Desai, T. y Barkel, D. (2008): “Syndromic colon cancer: Lynch syndrome and familial adenomatous polyposis”. En: Gastroenterol Clin N Am, 37: 47-72.
- Smith, J.C.; Schaffer, M.W.; Ballard, B.R. et al. (2013): “Adenocarcinoma after prophylactic surgery for familial adenomatous polyposis”. En: J Cancer Ther, 4(1): 260-270.
- Kastrinos, F. y Syngal, S. (2007): “Recently identified colon cancer predispositions: MYH and MSH6 mutations”. En: Semin Oncol, 34(5): 418-424.
- Kastrinos, F. y Syngal, S (2012): “Inherited colorectal cancer syndromes. Cancer J 2011;17(6):405-415Kerr SE, Thomas CB, Thibodeau SN, et al: APC Germline Mutations in Individuals Being Evaluated for Familial Adenomatous Polyposis A Review of the Mayo Clinic Experience with 1591 Consecutive Tests”. En: J Mol Diagn, 1-13.
- Peterlongo, P.; Mitra, N.; Sánchez, A. et al. (2006): “Increased frequency of disease causing MYH mutations in colon cancer families”. En: Carcinogenesis, 27(11): 2243-2249.
- Lefevre, J.H.; Rodríguez, C.M.; Mourra, N. et al. (2006): “Implications of MYH in colorectal polyposis”. En: Annals of Surg, 244(6): 874-880.
- Lubbe, S.J.; Di Bernardo, M.C.; Chandler, I.P. et al. (2009): “Clinical implications of the colorectal cancer risk associated with MUTYH mutation”. En: J Clin Oncol, 27(24): 3975-3980.
- Jenkins, M.A.; Croitoru, M.E.; Monga, N. et al. (2006): “Risk of colorectal cancer in monallelic and biallelic carriers of MYH mutations: a population based case family study”. En: Cancer Eidemiol Biomarkers Prev, 15: 312-314.
- Vasen, H.F.A.; Ghorbanoghli, Z.; Bourdeaut, F. et al. (2014): “Guidelines for surveillance of individuals with constitutional mismacht repair deficiency proposed by the European consortium ‘Care for CMMRD’”. En: J Med Genet, 51: 283-293.
- Lucci-Cordisco, E.; Risio, M.; Venesio, T. et al. (2013): “The growing complexity of the intestinal polyposis”. En: M J Med Gent A, 161A(11): 2777-2785.
- Valle, L. (2014): “Genetic predisposition to colorectal cancer: where we stand and future perspectives”. En: World J Gastroenterol, 20(29): 9828-9849.
- Gammon, A.; Jasperson, K.; Kohlmann, W. et al. (2009): “Hamartomatous polyposis syndromes”. En: Best Pract Res Clin Gastroenterol, 23(2): 219-231.
- Campos, F.G.; Figueiredo, M.N. y Real Martínez, C.A. (2015): “Colorectal cancer risk in hamartomatous polyposis syndromes”. En: World J Gastrointest Surg, 7(3): 25-32.
- Manfredi, M. (2010): “Hereditary Hamartomatous Polyposis Syndromes: Understanding the disease risk as children reach adulthood”. En: Gastroenterology and hepatology, 6(23): 185-196.
- Stojcev, Z.; Borun, P.; Hermann, J. et al. (2013): “Hmartomatous polyposis syndromes”. En: Hereditary Cancer in Clinical Practice, 11: 4.
- Cichy, W.; Klincewicz, Plawski, A. (2014): “Juvenile poluposis syndrome”. En: Arch Med Sci, 10(3): 570-577.
- Jelsig, A.M.; Qvistz, N.; Brusgaard, K. et al. (2014): “Hamartomatous polyposis syndromes: A review”. En: Orphanet J Rare Dis, 15(9):101.
- NCCN (s/f): “Guidelines Genetic/Familial High Risk assessment: Colorectal”. Disponible online en: <https://www.nccn.org>.
- Cao, X.; Eu, K.W.; Kumarasinghe, M.P. et al. (2006): “Mapping of hereditary mixed polyposis syndrome (HMPS) to chromosome 10q23 by genomewide high-density single nucleotide polymorphism (SNP) scan and identification of BMPR1A loss of function”. En: J Med Genet, 43: 13.
- Giardello, F.M. y Hamilton, S.R. (1997): “Hereditary mixed polyposis syndrome: a zebra or a horse dressed in pinstripes”. En: Gastroenterology, 112: 643-659.
- Thomas, H.J.W.; Whitelaw, S.C.; Cottrell, S.E. et al. (1996): “Genetic mapping of the hereditary mixed polyposis syndrome to chromosome 6q”. En: Am J Hum Genet, 58: 770-776.
- Whitelaw, S.C.; Murday, V.A.; Tomlinson, I.P.N. et al. (1997): “Clinical and molecular features of the hereditary mixed polyposis syndrome”. En: Gastroenterology, 112: 327-334.
- Bomparai, K.S.; Dekker, E.; Polak, M.M. et al. (2011): “A serrated colorrectal cancer pathway predominates over the classic WNT pathwayt in patients with hiperplastic polyposis syndrome”. En: Am J Pathol, 178(6): 2700-2706.
- Horii, J.; Kato, J.; Nagasaka, T. et al. (2013): “Development of invasive colon cancer with microsatellite instability in a patient with hiperplastic polyposis syndrome”. En: Jpn J Clin Oncol, 42(5): 451-454.
- Orlawska, J. (2013): “Serrated lesions and hyperplastic (serrated) polyposis relationship with colorectal cancer: classification and surveillance recommendations”. En: Gastrointestinal Endoscopy, 77(6): 858-871.
- Toyoshima, N.; Sakamoto, T.; Makazu, M. et al. (2015): “Prevalence of serrated polyposis syndrome and its association with synchronous advanced adenoma and lifestyle”. En: Molecula and Clinical Oncology, 3: 69-72.
- East, J.E.; Vieth, M. y Rex, D.K. (2015): “Serrated lesions in colorectal cancer screening: detection, resection, pathology and surveillance”. En: Gut, 64: 991-1000.
- East, J.E.; Saunders, B.P. y Jass, J.R. (2008): “Sporadic and Syndromic Hyperplastic Polyps and Serrated Adenomas of the Colon: Classification, Molecular Genetics, Natural History, and Clinical Management”. En: Gastroenterol Clin N Am, 37: 25-46.
- Edelstein, D.L.; Axilbund, J.E.; Hylind, K.M. et al. (2013): “Serrated polyposis: rapid and relentless development of colorectal neoplasia”. En: Gut, 62(3): 404-408.
- Rex, D.K.; Ahnen, D.J.; Baron, J.A. et al. (2012): “Serrated lesions of the colorectum: review and recommendations from an expert panel”. En: Am J Gastroenterol, 107990: 1315-1330.
- Hawkins, N.J.; Gorman, P.; Tomlinson, I.P.M. (2000): “Colorectal Carcinomas Arising in the Hyperplastic Polyposis Syndrome Progress through the Chromosomal Instability Pathway”. En: AJP, 157: 385-392.
- Win, A.K.; Walters, R.J.; Buchanan, D.D. et al. (2012): “Cancer risks for relatives of patients with serrated polyposis”. En: AM J Gastroenterol, 107(5): 770-778.
- Boparai, K.S.; Reitsma, J.B.; Lemmens, V. et al. (2013): “Increase colorectal cancer risk in first-degree relatives of patients with hyperplastic polyposis syndrome”. En: Gut, 59: 1222-1225.
- Rosty, C.; Hewett, D.G.; Brown, I.S. et al. (2013): “Serrated polyps of the large intestine: current understanding of diagnosis, pathogenesis and clinical management”. En: J Gastroenterol, 48: 287-302.
- Hazewinkel, Y.; López-Cerón, M.; East, J.E. et al. (2013): “Endoscopic features of sessile serrated adenomas: validation by international experts using high-resolution white-light endoscopy and narrow-band imaging”. En: Gastrointestinal Endoscopic, 77(6): 916-924.
- Guarinos, C.; Sánchez Fortun, C.; Rodríguez Soler, M. et al (2012): “Serrated polyposis syndrome: molecular, pathological and clinical aspects”. En: World J Gastroenterol, 18(20): 2452-2461.
- Roberts, A.; Nancarrow, D.; Clendenning, M. et al. (2011): “Linkage to chromosome 2q32.2-q33.3 in familial serrated neoplasia (Jass syndrome)”. En: Fam Cancer, 10: 245-254.
- Jasperson, K.W.; Kanth, P.; Kirchhoff, A.C. et al. (2013): “Serrated polyposis: colonic phenotype extracolonic features and familial risk in a large cohort”. En: Dis Colon Rectum, 56: 1211-1216.
VIGILANCIA POST CIRUGÍA DEL CÁNCER COLORRECTAL CON INTENCIÓN CURATIVA
A pesar de un tratamiento óptimo, entre un 30% y un 50% de los pacientes con cáncer colorrectal (CCR) desarrollará una recurrencia del tumor, y la mayoría de los recaídos morirá por la enfermedad. Aproximadamente, el 80% de las recurrencias se produce dentro de los tres años posteriores al tratamiento y un 95% dentro de los primeros 5 años.1,2
Después de 5 años la tasa de recurrencia es menor de 1,5% por año y después de los 10 años es menor a 0,5% por año. Por lo tanto, las recomendaciones de vigilancia están en gran medida circunscriptas a los primeros 5 años luego de la cirugía.
El seguimiento posterior a un tratamiento con intención curativa tiene como objetivos evaluar posibles complicaciones terapéuticas, descubrir recurrencias potencialmente curables y/o identificar tumores metacrónicos.
Existen muchas guías internacionales de práctica clínica con recomendaciones para la vigilancia de los pacientes después de un tratamiento curativo del CCR elaboradas en base a meta análisis y a estudios controlados y randomizados. Las guías principales publicadas son: la guía de National Comprehensive Cancer Network (NCCN, Clinical Practice Guidelines in Oncology: Colon Cancer-Rectal Cancer),3, 4, las guías de Cancer Care Ontario (CCO),5 avaladas por la American Society of Clinical Oncology (ASCO, Follow-Up Care, Surveillance Protocol, and Secondary Prevention Measures for Survivors of Colorectal Cancer: American Society of Clinical Oncology Clinical Practice Guideline Endorsement),6 la guía de European Society of Medical Oncology (ESMO, Clinical Practice Guidelines for Colon7 and Rectal Cancer)8 y, recientemente, la guía de American Society of Colon and Rectal Surgeons9 (ASCRS, Practice Guideline for the Surveillance of Patients After Curative Treatment of Colon and Rectal Cancer).
Las medidas de vigilancia sólo deben aplicarse a los pacientes que puedan ser susceptibles de resección de la recaída y que no tengan comorbilidades graves que impidan otra resección quirúrgica y/o una terapia sistémica.6-8 Los pacientes que no sean candidatos para la cirugía o que no sean candidatos para una terapia sistémica, debido a comorbilidades graves, no deberían realizar vigilancia. Por lo tanto, cuando se recomienda vigilancia deben considerarse fuertemente las comorbilidades del paciente, su edad, el nivel de su actividad, sus preferencias y su posibilidad de cumplimiento del programa de seguimiento elegido.
Las herramientas actuales para la vigilancia incluyen una combinación de la historia clínica, el examen físico, el laboratorio, las imágenes y la endoscopía en esquemas que varían en función del estadio inicial de la enfermedad, la situación clínica del paciente y la localización del tumor primario (colon versus recto).
La evaluación clínica debe incluir el interrogatorio acerca de antecedentes clínicos y quirúrgicos y el examen físico, dado que los síntomas pueden ser el primer signo de recurrencia en pacientes con CCR. Sin embargo, menos del 7% de los pacientes con recurrencia sintomática tienen enfermedad resecable. Por otro lado, los pacientes que tienen o han tenido CCR, especialmente aquellos que son mayores, tienen un mayor riesgo de enfermedades generales y de otros tumores malignos. Por lo tanto, las visitas al consultorio clínico proporcionan una oportunidad para un control general de su salud y para la detección de otros tumores primarios.
La determinación sérica de CEA debe realizarse en el contexto de las otras medidas de vigilancia. El CEA, aislado, sin otro examen auxiliar, no produjo beneficios en estudios individuales y demostró reducción de mortalidad solo en un meta-análisis.10
Sin embargo, la elevación del CEA, a menudo, es el primer signo de recurrencia. Un resultado positivo puede adelantarse entre un mes y medio y seis meses a la detección clínica y/o instrumental de la recurrencia con otros métodos diagnósticos. El monitoreo del CEA resulta efectivo aun en pacientes sin elevación preoperatoria del marcador. No hay evidencia de otros parámetros de laboratorio que resulten útiles para el seguimiento.7,8 Muchos grupos agregan al CEA la determinación de CA 19-9 sérico. Si bien parece contribuir a detectar recaídas, no hay guías que recomienden su uso rutinario.
La Tomografía Computada (TC) es útil para monitorear la presencia de lesiones metastásicas potencialmente resecables, principalmente en el hígado y el pulmón, sitios frecuentes de recurrencia sistémica del CCR.11 La TC de abdomen presenta mayor sensibilidad que la ecografía para la detección de metástasis hepáticas. La TC de tórax puede detectar recurrencia pulmonar (aproximadamente en el 20% de los pacientes constituye el primer sitio de recaída). Debe subrayarse que la resección de metástasis hepáticas o pulmonares determina una sobrevida de 25 a 50% a los 5 años, justificando fuertemente la detección temprana en esos órganos a través de programas de vigilancia adecuados. No hay datos a favor del seguimiento con Rx de tórax.7,8
La Videocolonoscopia (VCC) completa se recomienda antes del tratamiento quirúrgico curativo para identificar lesiones sincrónicas. Si por un tumor primario infranqueable o por otra causa no se pudiera hacer una VCC completa, esta debería realizarse dentro de los 3 a 6 meses posteriores a la cirugía. Ni la realización de esta VCC ni el cierre de una ostomía deberían postergar el comienzo del tratamiento adyuvante postoperatorio, en tiempo (entre 20 y 45 días después de la cirugía) si es que tal tratamiento corresponde.
La vigilancia con VCC se realiza principalmente para la identificación y la resección de pólipos metacrónicos y/o recurrencias intraluminales y/o segundos tumores primarios.
La tomografía por emisión de positrones (PET / TC) no se recomienda para la vigilancia de rutina.
La mayoría de las guías recomiendan la vigilancia, con las herramientas ya mencionadas, en los pacientes operados por CCR con enfermedad en estadios II y III y en pacientes seleccionados con CCR en estadio IV, sin evidencias de enfermedad después de un tratamiento con intención curativa. Las ventajas de un seguimiento más intensivo en estos pacientes se ha demostrado de forma prospectiva en varios estudios12-14 y en 3 meta-análisis de estudios controlados y randomizados diseñados para comparar los programas de vigilancia de baja y alta intensidad.11,15-17 Además, un informe de base poblacional indica que las tasas de resecabilidad y la sobrevida aumentaron en pacientes tratados por recurrencia local y metástasis a distancia del cáncer colorrectal, proporcionando de este modo un soporte para un seguimiento más intensivo post-tratamiento en estos pacientes.18
Persisten controversias sobre el papel de la vigilancia en pacientes en estadio I. CCO5 y ASCO6 no recomiendan la vigilancia por la falta de datos basados en evidencia. En cambio, NCCN3,4 y ASCRS9 consideran que un programa de vigilancia menos intensivo es apropiado. Los posibles daños incluyen la excesiva exposición a la radiación ante la repetición de las tomografías y el estrés psicológico asociado con las visitas de vigilancia y el riesgo de resultados falsos positivos de los diferentes estudios. Hay subgrupos de pacientes con estadio I que pueden tener alto riesgo de recaída en los que podría justificarse un programa de seguimiento, quizá de baja intensidad: aquellos con cáncer de recto luego de una escisión local, aquellos con pólipos malignos de alto riesgo que no realizaron una cirugía radical y los pacientes sometidos a cirugía radical con invasión linfovascular, márgenes positivos, pobremente diferenciados y/o con tumores T2.
Recomendaciones para el seguimiento de pacientes operados con intención curativa por un cáncer de colon:
- Efectuar historia clínica y examen físico cada 3 meses los primeros 2 años, luego cada 6 meses hasta completar los 5 años de la cirugía.
- Determinar CEA en sangre cada 3-6 meses los primeros 2 años, luego cada 6-12 meses hasta completar los 5 años de la cirugía.
- Hacer TC tórax, abdomen y pelvis anualmente durante 3 años (ASCO6/CCO5) o 5 años (NCCN3, ASCRS9).
- Efectuar VCC al año de la cirugía y luego repetirla a los 3 años y, posteriormente, cada 5 años, excepto que se encuentren lesiones que indiquen adenoma avanzado (pólipo velloso, pólipo mayor a 1 cm, displasia de alto grado). En este caso, se debe repetir en un año. En pacientes con cáncer de colon antes de los 50 años o en síndromes hereditarios, puede estar indicado realizar VCC más frecuentes (cada 12 a 24 meses).
Recomendaciones para el seguimiento de pacientes operados con intención curativa por un cáncer de recto:
- Efectuar historia clínica y examen físico cada 3 meses los primeros 2 años; luego, cada 6 hasta completar los 5 años de la cirugía.
- Determinar CEA en sangre cada 3-6 meses los primeros 2 años; luego, cada 6-12 meses hasta completar los 5 años de la cirugía.
- Hacer TC de tórax, abdomen y pelvis anualmente por 5 años. ASCO6/CCO5 recomiendan TC abdominal y tórax anualmente durante 3 años. En los pacientes de alto riesgo, la TC pelvis se debe considerar cada 6 a 12 meses durante los primeros 2-3 años, luego anualmente hasta los 5 años de la cirugía.
- Efectuar VCC al año de la cirugía y luego repetirla a los 3 años y, posteriormente, cada 5 años, excepto que se encuentren lesiones que indiquen adenoma avanzado (pólipo velloso, pólipo mayor a 1cm, displasia de alto grado). En este caso, se debe repetir en un año. En pacientes con cáncer de colon antes de los 50 años o en síndromes hereditarios, puede estar indicado realizar VCC más frecuentes (cada 12 a 24 meses).
- En pacientes resecados sin tratamiento RT/QT preoperatorio: rectosigmoideoscopía (RSC) cada 6 meses por 2 años. NCCN4 recomienda RSC con EUS o RMN cada 3-6 meses durante los primeros 2 años, luego cada 6 meses durante 5 años (en los pacientes tratados con resección transanal solamente).
El seguimiento rutinario en CCR con CEA y TC de tórax, abdomen y pelvis no se recomienda pasados los 5 años del diagnóstico.
Las recomendaciones ante niveles elevados de CEA después de la resección, según recomendaciones de la NCCN, deben incluir examen físico, VCC, TC de tórax, abdomen y pelvis, y la consideración de PET/TC. Si los resultados de los estudios por imágenes, incluyendo PET, son normales ante un aumento del CEA, repetir la exploración con TC cada 3-4 meses hasta que se identifique la enfermedad o los niveles de CEA se estabilicen o disminuyan.
La vigilancia de los pacientes operados con intención curativa por un CCR incluye un programa de prevención secundaria (vacunas, medidas de prevención de otras enfermedades; detección temprana a través de tamizaje periódico para un segundo cáncer primario como cáncer de mama, cáncer de cuello uterino o cáncer de próstata).
La evidencia indica que ciertas medidas preventivas relacionadas con el estilo de vida, dejar de fumar, mantener un índice de masa corporal saludable, practicar ejercicio regularmente, y ciertos hábitos dietéticos se asocian con mejores resultados en sobrevida y en calidad de vida.
Un estudio de pacientes en estadios I a III de cáncer colorrectal mostró una asociación entre el aumento de la actividad física y menores tasas de mortalidad específica por CCR y menor mortalidad global.19 Además, una reciente revisión sistemática y un meta-análisis sugieren que la actividad física realizada antes o después del diagnóstico disminuye la mortalidad por CCR.20 Se observaron resultados similares en otro reciente meta-análisis de estudios prospectivos.21
El mecanismo de los beneficios de la actividad física y de la dieta probablemente está relacionado con el peso corporal, dado que el exceso de peso es un factor de riesgo para el CCR, que es modificable a través de cambios del estilo de vida.22 Análisis recientes confirman el aumento del riesgo de recurrencia y muerte en pacientes obesos.23
Dejar de fumar tiene un impacto clínico en estos pacientes, dado que continuar con el hábito después del diagnóstico de cáncer colorrectal se ha relacionado con una baja sobrevida.24
La evidencia de estudios observacionales sugiere que una dieta baja en grasas y alta en fibra puede tener un efecto protector contra la recurrencia y la progresión del CCR.22 El vínculo entre las carnes rojas y procesadas y la mortalidad en los sobrevivientes de CCR ha sido apoyado por datos recientes: pacientes con alto consumo tuvieron un mayor riesgo de mortalidad específica por CCR que aquellos con una ingesta baja (RR, 1.79; IC del 95%, 1.11-2.89).25
También hay alguna evidencia de que una mayor ingesta de productos lácteos puede estar asociada con un menor riesgo de muerte en pacientes con estadios I, II o III de CCR.26
Otras intervenciones, incluyendo la administración de vitamina D, el uso regular de aspirina o un inhibidor de la ciclooxigenasa, requieren más estudios para obtener una evidencia que justifique su uso rutinario para la prevención secundaria.
BIBLIOGRAFÍA
- Sargent, D.J.; Patiyil, S.; Yothers, G. et al. (2007): “End points for colon cancer adjuvant trials: Observations and recommendations based on individual patient data from 20,898 patients enrolled onto 18 randomized trials from the ACCENT Group”. En: J Clin Oncol, 25: 4569-4574.
- Seo, S.I.; Lim, S.B.; Yoon, Y.S. et al. (2013): “Comparison of recurrence patterns between ≤5 years and >5 years after curative operations in colorectal cancer patients”. En: J Surg Oncol., 108(1): 9-13.
- NCCN (s/f): “Clinical Practice Guidelines in Oncology (NCCN Guidelines): Rectal Cancer. Version 1.2016”. Disponoible online en: <http://www.nccn.org>.
- NCCN (s/f): “Clinical Practice Guidelines in Oncology (NCCN Guidelines): Rectal Cancer. Version 1.2016”. Disponoible online en: <http://www.nccn.org>.
- Earle, C.; Annis, R.; Sussman, J. et al. (s/f): “Follow-up care, surveillance protocol, and secondary prevention measures for survivors of colorectal cancer”. Disponible online en: <https://cancercare.on.ca>.
- Meyerhardt, J.A.; Mangu, P.B.; Flynn, P.J. et al. (2013): “American Society of Clinical Oncology. Follow-up care, surveillance protocol, and secondary prevention measures for survivors of colorectal cancer: American Society of Clinical Oncology clinical practice guideline endorsement”. En: J Clin Oncol., 31: 4465-4470.
- Schmoll, H.J.; Van Cutsem, E.; Stein, A. et al. (2012): “ESMO Consensus Guidelines for management of patients with colon and rectal cancer. A personalized approach to clinical decision making”. En: Ann Oncol., 23(10): 2479-2516.
- Glimelius, B.; Tiret, E.; Cervantes, A. y Arnold, D. (2013): “Rectal cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up”. En: Ann Oncol. 24 Suppl 6: 81-88.
- Steele, S.R.; Chang, G.J.; Hendren, S. et al. (2015): “Practice Guideline for the Surveillance of Patients After Curative Treatment of Colon and Rectal Cancer. Clinical Practice Guidelines Committee of the American Society of Colon and Rectal Surgeons”. En: Dis Colon Rectum., 58(8): 713-725.
- Chau, I.; Allen, M.J.; Cunningham, D. et al. (2004): “The value of routine serum carcinoembryonic antigen measurement and computed tomography in the surveillance of patients after adjuvant chemotherapy for colorectal cancer”. En: J Clin Oncol, 22: 1420-1429.
- Desch, C.E.; Benson, A.B.; Somerfield, M.R. et al. (2005): “American Society of Clinical Oncology. Colorectal cancer surveillance: 2005 update of an American Society of Clinical Oncology practice guideline”. En: J Clin Oncol., 23: 8512-8519.
- Pietra, N.; Sarli, L.; Costi, R.; Ouchemi, C. et al. (1998): “Role of follow-up in management of local recurrences of colorectal cancer”. En: Dis Colon Rectum, 41: 1127-1133.
- Rodríguez-Moranta, F; Saló, J.; Arcusa, A. et al. (2006): “Postoperative surveillance in patients with colorectal cancer who have undergone curative resection: a prospective, multicenter, randomized, controlled trial”. En: J Clin Oncol., 24: 386-393.
- Secco, G.B.; Fardelli, R.; Gianquinto, D. et al. (2002): “Efficacy and cost of risk-adapted follow-up in patients after colorectal cancer surgery: a prospective, randomized and controlled trial”. En: Eur J Surg Oncol., 28: 418-423
- Jeffery, M.; Hickey, B.E.; Hider, P.N. (2007): “Follow-up strategies for patients treated for non-metastatic colorectal cancer”. En: Cochrane Database Syst Rev., 24(1): CD002200.
- Figueredo, A.; Rumble, R.B.; Maroun, J. et al. (2003): “Follow-up of patients with curatively resected colorectal cancer: a practice guideline”. En: BMC Cancer, 3: 26.
- Renehan, A.G.; Egger, M.; Saunders, M.P. y O’Dwyer, S.T. (2002): “Impact on survival of intensive follow up after curative resection for colorectal cancer: systematic review and meta-analysis of randomised trials”. En: BMJ. 324(7341): 813.
- Guyot, F.; Faivre, J.; Manfredi, S. et al. (2005): “Time trends in the treatment and survival of recurrences from colorectal cancer”. En: Ann Oncol., 16(5): 756-761.
- Meyerhardt, J.A.; Giovannucci, E.L.; Ogino, S. et al. (2009): “Physical activity and male colorectal cancer survival”. En: Arch Intern Med., 169(22): 2102-2108.
- Schmid, D. y Leitzmann, M.F. (2014): “Association between physical activity and mortality among breast cancer and colorectal cancer survivors: a systematic review and meta-analysis”. En: Ann Oncol., 25(7): 1293-1311.
- Je, Y.; Jeon, J.Y.; Giovannucci, E.L. y Meyerhardt, J.A. (2013): “Association between physical activity and mortality in colorectal cancer: A meta- analysis of prospective cohort studies”. En: Int J Cancer, 133(8): 1905-1913.
- Davies, N.J.; Batehup, L. y Thomas, R. (2011): “The role of diet and physical activity in breast, colorectal, and prostate cancer survivorship: a review of the literature”. En: Br J Cancer, 105 Suppl 1:S52-73.
- Sinicrope, F.A.; Foster, N.R.; Sargent, D.J. et al. (2010): “Obesity Is an Independent Prognostic Variable in Colon Cancer Survivors”. En: Clin Cancer Res, 16(6); 1884-1893.
- Walter, V.; Jansen, L.; Hoffmeister, M. y Brenner, H. (2014): “Smoking and survival of colorectal cancer patients: systematic review and meta-analysis”. En: Ann Oncol., 25(8): 1517-1525.
- McCullough, M.L.; Gapstur, S.M.; Shah, R. et al. (2013): “Association between red and processed meat intake and mortality among colorectal cancer survivors”. En: J Clin Oncol., 31(22): 2773-2782.
- Yang, B.; McCullough, M.L.; Gapstur, S.M. et al. (2014): “Calcium, Vitamin D, Dairy Products, and Mortality Among Colorectal Cancer Survivors: The Cancer Prevention Study-II Nutrition Cohort”. En: J Clin Oncol., 32(22): 2335-2343.
VIGILANCIA EN LA ENFERMEDAD INFLAMATORIA INTESTINAL
Los pacientes con enfermedad inflamatoria intestinal (EII) presentan un mayor riesgo de cáncer colorrectal que la población general. Es importante resaltar que los enfermos con colitis ulcerosa (CU) poseen un riesgo similar al que tienen los portadores de la enfermedad de Crohn (EC), considerando una duración y una extensión del compromiso colónico similares.
La vigilancia colonoscópica en pacientes con EII tiene como objetivo detectar una lesión pre neoplásica, prevenir la potencial progresión al cáncer de colon e identificar un cáncer invasor en estadio curable.
Existen diferentes publicaciones con recomendaciones sobre vigilancia de cáncer colorrectal en este grupo de pacientes, con diferentes diseños y resultados controvertidos. En el año 2004, la agrupación Crohn’s and Colitis Foundation of America organizó una convención sobre “Cáncer de colon en Enfermedad inflamatoria intestinal”, donde un grupo de expertos internacionales efectuó una redefinición de las recomendaciones para pesquisa y vigilancia del cáncer colorrectal que se habían efectuado en el año 2000 que fue posteriormente publicada.1
Factores de riesgo a tener en cuenta en el diseño de un programa de vigilancia en EII
El tiempo de evolución y la extensión de la lesión colónica son las variables demostradas en diferentes estudios como factores de riesgo independiente.
Según un meta-análisis de Eaden y colaboradores,2 la prevalencia global del cáncer colorrectal en la colitis ulcerosa es del 3,7%. El riesgo de desarrollar cáncer de colon en los pacientes con colitis ulcerosa está estimado en un 2% a los 10 años de evolución de la enfermedad, en un 8% a los 20 años y en un 18% a los 30 años. Ransohoff3 estimó un riesgo anual de 0,5 a 1% a partir de los 8 a 10 años de evolución.
Diferentes autores reportaron un incremento anual constante desde los 15 años de enfermedad; observándose la mayor parte de los cánceres en las pancolitis, con una prevalencia del 5,4%.2
El riesgo de cáncer en las proctitis no se ve incrementado, mientras que en las colitis izquierdas es intermedio, observándose 5 a 10 años después de la colitis extensa.1,4 Hay distintas metodologías para definir la extensión. En un estudio donde se evaluó la extensión mediante colon por enema y colonoscopia evidenció en la proctitis un riesgo de 1,7 (IC 95%: 0,8-3,2); en la colitis izquierda de 2,8 (IC 95%: 1,6-4,4) y en la pancolitis de 14,8 (IC 95%: 11,4-18,9).5 Debido a estos hallazgos, se recomendó el inicio de la vigilancia a los 8-10 años en la enfermedad extensa y a los 15-20 años en la izquierda.6,7
Existen discrepancias según la forma de clasificación de la extensión, por criterios endoscópicos o histológicos (desacuerdo: del 22% al diagnóstico y del 40% a los 4 años).8 En la pancolitis la discordancia es menor, no obstante un 13% de los casos se clasificaría como colitis extensa sólo por la histología; inversamente, un 37% de pacientes tenían histología normal en áreas con sospecha endoscópica de lesión.9
Una revisión realizada en piezas quirúrgicas detectó displasia y cáncer en áreas de colitis microscópicas, localizadas proximalmente al segmento comprometido macroscópicamente.
Colangitis esclerosante primaria
Diferentes estudios reportaron un mayor riesgo de cáncer colorrectal en los pacientes con colitis ulcerosa asociada a colangitis esclerosante primaria con respecto a aquellos con colitis ulcerosa sin dicha asociación.11, 12 Un estudio reportó un riesgo acumulativo absoluto del cáncer o displasia en la colangitis esclerosante primaria asociada a colitis ulcerosa del 9% después de 10 años, 31% después de 20 años y 50% después de 25 años de enfermedad colónica.12
En el Hospital de Gastroenterología de Buenos Aires se realizó un estudio de pacientes con colitis ulcerosa con y sin colangitis esclerosante primaria asociada, detectando un mayor riesgo para cáncer colorrectal en dicha asociación (OR: 24,6; IC 95%: 2,9-205,4).13
Una publicación de la Mayo Clinic llevada a cabo por Loftus y cols.14,15 no observó mayor riesgo de cáncer en este grupo de pacientes en comparación con el resto de las colitis ulcerosas, aunque describen una prevalencia aumentada en los pacientes trasplantados por el mismo diagnóstico. Las normativas de vigilancia actuales establecen recomendaciones especiales para este subgrupo de pacientes, que se incluyen en un grupo de alto riesgo para desarrollar un cáncer colorrectal.
Historia familiar de cáncer colorrectal
Existen reportes de un aumento del riesgo en estos casos, habiéndose estimado en algunos estudios un riesgo duplicado si existe una antecedente familiar de primer grado y de nueve veces más si este familiar es menor de 50 años.16,17 Este dato debería documentarse para mejorar la información y actuar basándose en el criterio clínico.
Grado de actividad endoscópica e histológica
Si bien está reportada la correlación entre displasia y severidad endoscópica-histológica, debe tenerse en cuenta que aquella puede también aparecer en pacientes con enfermedad quiescente. La interpretación histológica es más sencilla cuando la enfermedad está en remisión, ya que algunos rasgos histológicos de la enfermedad activa pueden ser confundidos con displasia.
En líneas generales, la actividad de la enfermedad puede generar incertidumbre en pocos casos y la confiabilidad de la interpretación está en relación con la experiencia del patólogo. Ello implica que, en el caso de enfermedad activa, no se justifica diferir los estudios demasiado tiempo alterando el programa de vigilancia con el fin de incrementar la exactitud diagnóstica. No obstante, si la demora no es significativa, es aceptable intentar previamente un tratamiento para reducir la inflamación.
Otros factores predisponentes
El inicio de la enfermedad a edad temprana, la presentación clínica crónica continua y el persistente proceso inflamatorio se describen como factores predisponentes para un mayor riesgo de cáncer.1,4,18 Sin embargo, debido a las controversias existentes, estos aspectos no modifican las recomendaciones de vigilancia establecidas.19,20
Agentes farmacológicos para la prevención del cáncer colorrectal en la EII.
La quimioprevención representa un interesante y promisorio abordaje para intervenir tempranamente en la secuencia carcinogénica, antes del desarrollo de displasia o carcinoma, evitando la necesidad de la colectomía. El agente ideal para la quimioprevención debería tener un mecanismo de acción bien comprendido, ser seguro, efectivo y aceptado por los pacientes.
Diferentes estudios han descripto el rol de los aminosalicilatos en la quimioprevención. En el estudio caso-control llevado a cabo por Eaden y col.,21 el uso de aminosalicilatos a dosis de 1,2 g/d o mayor estuvo asociado con una reducción del 75% de riesgo de cáncer, aunque el papel protector de esta droga no ha sido universalmente reportado.
Un meta-análisis sobre el uso de 5-asa y el riesgo de displasia o cáncer fue realizado por Velayos y col.22 incluyendo 1.932 pacientes, con 334 casos de CCR y 140 casos de displasia. El resultado de su análisis reveló un OR 0,51 (CI 95%: 0,38-0,69) para el desarrollo de displasia o cáncer entre los pacientes con EII que usaban regularmente aminosalicilatos.
El estudio de cohorte Cesame “Cancers et sur-risque associé aux maladies inflammatoires chroniques intestinales en France”23 confirmó el papel preventivo del 5-asa en la colitis ulcerosa, en un estudio de 19.486 pacientes con EII, de los cuales el 40% eran colitis ulcerosas o EII no clasificada. El mencionado estudio francés no demostró el mismo efecto preventivo para la azatioprina. Tampoco se detectó en estudios previos que la azatioprina pueda incrementar el riesgo de cáncer.24
Un meta-análisis sobre tiopurinas y riesgo de cáncer colorrectal no observó efecto protector significativo de neoplasia colorrectal en EII.25 Sin embargo, existen publicaciones que reportan un rol protector de la Azatioprina.26 Debido a estas controversias, se requieren más estudios que confirmen estas observaciones.
El ácido fólico ha mostrado un efecto protector en el CCR. Este hallazgo, sumado a la observación de que muchos pacientes con EII pueden tener déficit de folatos, fue lo que motivó el interés de examinar su rol en la quimioprevención. Aunque los resultados no fueron estadísticamente significativos, se observó una tendencia hacia un efecto protector del ácido fólico. Hay algunas evidencias que podría disminuir el riesgo de cáncer, pero no son categóricas, y los factores como el tiempo de tratamiento y dosis no son equiparables entre los estudios.27,28
El ácido ursodesoxicólico (UDCA) fue evaluado en pacientes con colitis ulcerosa y colangitis esclerosante primaria, en los que se observó una disminución del riesgo de cáncer de colon.29,30
Procedimientos endoscópicos para la pesquisa y vigilancia en la EII
Conceptos generales de pesquisa/vigilancia
- Primer examen (pesquisa): se define como el primer examen colonoscópico en paciente con colitis ulcerosa o enfermedad de Crohn, con el objetivo de detectar displasia o cáncer colorrectal.
- Exámenes de vigilancia: colonoscopias subsiguientes realizadas periódicamente con el mismo propósito.1
Concepto de Displasia: se define como cambios neoplásicos inequívocos de la mucosa intestinal.
La displasia puede ser un marcador de que el cáncer está presente o está próximo en un futuro cercano. Éste es el motivo de su búsqueda mediante el seguimiento colonoscópico.31,32
Clasificación histológica e interpretación: se la clasifica en negativa, indefinida y positiva; esta última, subclasificada en alto grado y bajo grado.32 La indefinida puede optativamente sub clasificarse en probablemente positiva, probablemente negativa o desconocida. La displasia de bajo grado presenta cambios nucleares confinados en la porción basal de las criptas; en la displasia de alto grado tales cambios son más prominentes y se extienden a la superficie.32
El proceso inflamatorio puede ocasionalmente provocar cambios epiteliales que simulan displasia; por tal motivo, los estudios para la detección de la displasia deberían realizarse en los períodos de quiescencia.7,32
La presencia de un cáncer colorrectal en la pieza quirúrgica se asocia a displasia en el 90% de los casos. La detección de displasia de alto grado en mucosa plana observada en la colonoscopia de pesquisa se asocia con cáncer sincrónico en la pieza quirúrgica en el 42 al 67% de los casos. Si la detección es durante la vigilancia, la prevalencia de malignidad es del 32%.33,34
El hallazgo de una displasia requiere la confirmación de por al menos otro patólogo experto, ya que existe un grado considerable de variación interobservador. Por lo general, hay un mayor acuerdo en la displasia negativa y la displasia de alto grado, con mayor dificultad para discriminar entre displasia de alto y de bajo grado.35,36
Existen datos contradictorios respecto a la historia natural de la displasia de bajo grado. Bernstein34 reporta una transformación en displasia de alto grado o displasia asociada a lesión o masa (DALM) en el 29% de los casos, con una evolución a cáncer en el 8,1%, y que un 16% se negativiza. La experiencia del Hospital St. Marks de Londres37 demostró que la displasia de bajo grado en el 54% era predictiva de displasia de alto grado o cáncer en 5 años, experiencia equiparable a la descripta por Ullman y col.,38 que observaron tal evolución en el 53% en el mismo período, encontrándose un cáncer estadio 1 en un 18% de los pacientes colectomizados dentro de los 6 meses de la detección de la displasia de bajo grado, y estadio 2-3 en un 20% en los que se negaron a la vigilancia. Estos datos difieren con los estudios de Befrits y col.39 y de Lim y col.,40 que observa porcentajes de 3% y 10% de progresión a displasia de alto grado o DALM y displasia de alto grado o cáncer respectivamente a 10 años.
La displasia de cualquier grado asociada a lesión o masa (DALM) ha sido tradicionalmente considerada indicación de cirugía. En la actualidad, se acepta que no todos los tipos de displasia polipoide en pacientes con colitis ulcerosa tienen el mismo significado:
- Displasia en un “pólipo adenoma-like”: es factible de resecar con polipectomía, estrechando la vigilancia.41,42
- Displasia de cualquier tipo en un pólipo no resecable endoscópicamente: es indicación quirúrgica.1
- Displasia en mucosa plana: su manejo dependerá de la clasificación histológica. La displasia de alto grado, de confirmarse con un nuevo examen, es indicación de colectomía ya que su hallazgo se asocia con la presencia de un cáncer en el 30-40% de los casos.
Recomendaciones de vigilancia en la colitis ulcerosa
La colonoscopia de pesquisa para descartar displasia o cáncer de colon se recomienda efectuarla 8 a 10 años luego del inicio de los síntomas compatibles con colitis ulcerosa. La enfermedad se clasifica en extensa (mayor al ángulo esplénico), izquierda (no sobrepasa el ángulo esplénico con compromiso al menos histológico mayor o igual a 35 cm), distal o proctosigmoiditis (recto con o sin lesión en sigmoides).1 La extensión se define por endoscopia e histología, eligiendo el método que demuestre el compromiso más extenso. La misma puede cambiar con el tiempo, por esto se debe continuar con la estrategia de vigilancia basada en la máxima extensión documentada.
Recomendaciones anteriores:
El intervalo de 2 años entre las colonoscopias de vigilancia está basado en el tiempo reportado para desarrollo de un cáncer después de una colonoscopia.37,40 A partir de los 20 años de evolución, se recomienda realizar vigilancia cada 1 a 2 años, debido al incremento en el riesgo de cáncer de colon.41
Los pacientes con CEP deben ser seguidos con colonoscopias anuales.
Otros factores de riesgo –como historia familiar de cáncer colorrectal, pólipos inflamatorios, estenosis y una actividad persistente– pueden requerir intervalos de vigilancia más cortos.
Los pacientes con proctosigmoiditis, por tener mínimo o ningún riesgo de cáncer asociado, deben manejarse con las recomendaciones de prevención de cáncer colorrectal similares a la población general. En algunos pacientes se observa la presencia de un parche cecal eritematoso con inflamación microscópica, pero de acuerdo a las evidencias disponibles no se debe alterar esta recomendación.
Las guías británicas para pesquisa de cáncer de colon en EII, publicadas en 2010,43 recomiendan comenzar la vigilancia 10 años después del inicio de los síntomas. Clasifica el riesgo de desarrollar cáncer de colon en bajo, medio y alto.
Bajo riesgo:
- Colitis ulcerosa extensa en remisión.
- Enfermedad de Crohn extensa en remisión.
- CU izquierda con inflamación o EC con afectación menor al 50% de la superficie.
Riesgo intermedio:
- CU o EC extensa con actividad endoscópica o histológica leve.
- Pólipos post inflamatorios.
- Historia de cáncer de colon en familiar de 1° grado mayor de 50 años.
Riesgo alto:
- CU o colitis de Crohn con actividad endoscópica/histológica moderada a severa.
- CEP (aún después del trasplante).
- Estenosis de colon en los últimos 5 años.
- Displasia de cualquier grado en los últimos 5 años.
- Historia de cáncer de colon en familiar de 1° grado menor de 50 años.
La vigilancia propuesta en el grupo de bajo riesgo es colonoscopia cada 5 años; en el de riesgo intermedio, es colonoscopia cada 3 años; y en el de riesgo alto, la indicación es colonoscopia anual.
Respecto de la vigilancia en los pacientes colectomizados con reservorio ileal, las recomendaciones en estas guías clasifican a los pacientes en alto y bajo riesgo. Pacientes de alto riesgo serían aquellos con displasia rectal previa, displasia o cáncer de colon en la cirugía, CEP o mucosa tipo C en el pouch (atrofia severa). La vigilancia endoscópica debe ser anual. En los pacientes de bajo riesgo (aquellos donde no existe ninguna situación anteriormente descripta) la evaluación es cada 5 años.
Recomendaciones actuales
La vigilancia mediante biopsias escalonadas como estándar fue establecido previo a la aparición y disponibilidad de endoscopios de alta resolución y nuevas técnicas de inspección de la mucosa. Rubin y col.44 estimaron en el año 1992 que para obtener un porcentaje adecuado de detección de lesiones displásicas en mucosa plana, debían obtenerse entre 33 y 64 biopsias aleatorias en la colonoscopía, sin embargo, en la actualidad se conoce que con este número de biopsias, el porcentaje de mucosa evaluado es muy bajo. En una encuesta realizada en EEUU a 300 profesionales, aproximadamente el 50% de los gastroenterólogos cumple con este número de biopsias. La tasa de detección de displasia mediante este método es baja, estimada en 0,2%. Si la búsqueda de displasia es mediante biopsias dirigidas la tasa de detección aumenta a 23%.45 Es reconocido el hecho de que la mayoría de las lesiones displásicas son visibles para la endoscopia.46
Es importante destacar que el uso de las nomenclaturas DALM (traducido como lesión o masa asociada a displasia), lesiones tipo adenoma o no adenomatosas actualmente se ha abandonado. Actualmente se utiliza la nomenclatura propuesta por el consenso internacional SCENIC47 para informar los hallazgos durante la vigilancia colonoscópica en los pacientes con enfermedad inflamatoria intestinal, modificada de la clasificación de París48 (tabla 40).
Si bien existe acuerdo entre expertos que los pacientes con EII deben ser sometidos a vigilancia, la implementación y la metodología de ella es motivo de desacuerdo, lo cual se refleja en la diferencia de las recomendaciones entre las distintas guías que existen hasta la fecha.
Recomendaciones internacionales de pesquisa del cáncer colorrectal en la enfermedad inflamatoria intestinal
- Declaración de posición médica en el diagnóstico y manejo de neoplasias colorrectales en EII. Asociación Americana de Gastroenterología, AGA 2010.49
- La técnica de vigilancia colonoscópica en pacientes con EII debe incluir biopsias extensas de todos los segmentos anatómicos de la mucosa colorrectal.
- Aunque no hay datos adecuados disponibles para recomendar un intervalo de vigilancia óptimo, se sugieren intervalos de 1 a 3 años.
- La inspección cuidadosa de la mucosa junto a un número suficiente de biopsias deberían obtenerse de todos los segmentos anatómicos del colon.
-
Consenso Europeo basado en evidencia para endoscopia en Enfermedad Inflamatoria Intestinal, ECCO, 2013.50
La vigilancia debe realizarse en todos los pacientes excepto en aquellos con proctitis o colitis de Crohn comprometiendo sólo un segmento colónico. Al no existir evidencia clara de los intervalos de vigilancia, intervalos individualizados basados en la estratificación de riesgo se recomiendan: - Alto riesgo definido como estenosis o displasia detectada en los últimos cinco años, colangitis esclerosante primaria, colitis extensa con inflamación activa severa o historia familiar de CCR en un familiar de primer grado antes de los 50 años: colonoscopía anual.
- Riesgo intermedio definido como colitis extensa con inflamación activa leve a moderada, presencia de pseudopólipos o historia familiar de CCR en familiar de primer grado mayor de 50 años: colonoscopia cada 2 a 3 años.
- Pacientes que no cumplan criterios de riesgo alto ni intermedio: colonoscopía cada 5 años.
La vigilancia colonoscópica debe ser efectuada por cromoendoscopia pancolónica con azul de metileno o índigo carmín, con biopsias dirigidas de cualquier lesión visible. Si no existe la experiencia apropiada en cromoendoscopia, se deben realizar biopsias escalonadas (4 cada 10 cm.); sin embargo, esto es inferior a la cromoendoscopia en la detección de lesiones neoplásicas.
- Posición del Consenso Internacional SCENIC para vigilancia y manejo de la displasia en EII, Sociedad Americana de Endoscopia Gastrointestinal (ASGE), 2015.47
Declaración 1: Cuando se realice vigilancia con colonoscopio de luz blanca, se recomienda alta definición por sobre definición estándar.
Declaración 2: Cuando se realice vigilancia con colonoscopio de definición estándar, se recomienda cromoendoscopia por sobre colonoscopía con luz blanca.
Declaración 3: Cuando se realice vigilancia con colonoscopio de alta definición, se recomienda cromoendoscopia por sobre colonoscopía con luz blanca.

Vemos entonces que las guías de la AGA recomiendan una vigilancia uniforme en intervalos y basada en biopsias escalonadas, versus las guías europeas que recomiendan intervalos según estratificación de riesgo y como primera opción el uso de cromoendoscopia y biopsias dirigidas. Se puede ver, como, en las últimas guías disponibles (ASGE), ya se recomienda en forma definitiva el uso de cromoendoscopia por sobre cualquier otra técnica.
Manejo de hallazgos anormales en la vigilancia de la colitis ulcerosa
Cualquier examen con al menos una biopsia categorizada como “indefinida para displasia”, “displasia de bajo grado”, “displasia de alto grado”, o “adenocarcinoma” es considerado un hallazgo anormal.
La decisión de la colectomía debe ser evaluada basándose en la eficiencia de la vigilancia realizada, involucrando la calidad de la preparación, la suficiencia de las biopsias, y la presencia o ausencia de inflamación activa, que ocasionalmente puede dificultar la interpretación de las biopsias. En los últimos años se ha aceptado que el manejo de la displasia polipoide difiere de la displasia en mucosa plana, siendo importante el concepto de factibilidad de resección.
Presencia de displasia
Displasia de bajo grado en mucosa plana: en primer lugar, se requiere la revisión por un patólogo experto en estas enfermedades.
Debido a la variabilidad de los datos de los estudios, el manejo es controvertido. El hallazgo de la displasia durante la vigilancia no representa el mismo riesgo de progresión a displasia de alto grado o cáncer que cuando se presenta en el examen de pesquisa.34,51 El riesgo de progresión a displasia de alto grado o cáncer a 5 años varía de estudios con valores superiores al 50% versus otros con una presentación infrecuente.37,40 En pacientes con displasia de bajo grado, puede estar presente un cáncer no reconocido en el 20% de los pacientes que son sometidos a cirugía a corto plazo.34,38 Por consiguiente, las opciones deben discutirse con los pacientes, ofreciéndoles una colectomía profiláctica, por la posibilidad de un adenocarcinoma sincrónico, pero también informando las posibles complicaciones de la proctocolectomía restaurativa.
Cuando la displasia bajo grado es multifocal (2 o más biopsias de un mismo examen de vigilancia) o repetitiva (2 o más exámenes con al menos un foco de displasia de bajo grado) tiene indicación de colectomía total profiláctica.38
Si la elección ante la displasia de bajo grado es continuar con la vigilancia, se recomienda repetir la colonoscopia en 3 meses (máximo 6 meses) con suficiente toma de biopsias, independientemente de la uni o multifocalidad de la displasia de bajo grado en mucosa plana. Si este examen resulta negativo para displasia, lo aconsejable es continuar con estudios frecuentes (cada 6 meses) y un adecuado muestreo histológico.38,51
Si el diagnóstico es el de displasia indefinida en mucosa plana, es necesaria la revisión por un patólogo experto y, de ser ratificado el diagnóstico, se requiere una nueva colonoscopia en 3 a 6 meses.
La displasia de alto grado en mucosa plana deberá ser confirmada por un patólogo experto. En caso de ratificarse el diagnóstico, la conducta es la realización de una proctocolectomía, dada la elevada proporción de adenocarcinomas sincrónicos y metacrónicos.34,37
Lesiones elevadas (pólipos) con displasia
Con respecto a las lesiones polipoideas dentro de las áreas de colitis, si es factible se debe efectuar polipectomia y tomar 4 biopsias de la mucosa adyacente a la misma y enviarlas para su estudio en forma separada.41,42 Si la polipectomia es completa y las biopsias adyacentes negativas para displasia, se requiere un nuevo examen en 6 meses. Si éste es negativo, puede continuarse con la vigilancia de rutina.
Si la lesión polipoide no es resecable endoscópicamente, o si la mucosa circundante presenta algún grado de displasia, está indicada una proctocolectomía por el riesgo aumentado de cáncer colorrectal sincrónico.
La alternativa de realizar una colectomía segmentaria no se ha evaluado en la literatura y debe limitarse a pacientes con un riesgo aumentado para la colectomía.
Los pólipos encontrados en áreas del colon libres de enfermedad, pueden manejarse según las recomendaciones de los adenomas esporádicos.52
Recomendaciones para pacientes con colitis de Crohn
En los pacientes con EC con compromiso sólo del intestino delgado, las recomendaciones para la prevención del cáncer de colon son las mismas que para la población general.52
En los enfermos con colitis de Crohn, el riesgo es similar a la colitis ulcerosa con la misma extensión de compromiso colónico y tiempo de evolución.53
Se conoce poco sobre quimioprevención.54
Recomendaciones para la vigilancia en pacientes con EC
El comienzo de la vigilancia endoscópica aconsejado es 8 a 10 años después del inicio de los síntomas.
Arbitrariamente está indicada la vigilancia a pacientes que tienen comprometido más de la tercera parte del colon al examen endoscópico (no se consensuó por el compromiso histológico del colon).54
En la colangitis esclerosante primaria asociada la recomendación es de colonoscopia anual, debido al aumento del riesgo de cáncer colorrectal.
Otros factores de riesgo, como historia familiar de cáncer, debut temprano de la enfermedad, grado de actividad endoscópica e histológica, se toman en consideración en algunas guías para modificar las recomendaciones de vigilancia.
En el caso de displasia o cáncer en una enfermedad segmentaria, no hay sustento bibliográfico para decidir el tipo de cirugía, una resección segmentaria o una proctocolectomía.
La eficiencia de un programa de vigilancia depende de varios factores, como son: la selección de los pacientes, la práctica colonoscópica, la preparación del colon, el manejo de las biopsias y la comunicación entre los clínicos, patólogos y endoscopistas ante hallazgos histológicos anormales.
Se recomienda la conveniencia de efectuar los exámenes de pesquisa o de vigilancia en períodos de inactividad de la enfermedad, ya que en algunos casos puede dificultar la interpretación histológica.
La práctica colonoscópica y el manejo de las muestras son similares a los de la colitis ulcerosa.
Manejo de situaciones clínicas especiales
Pólipos post inflamatorios:
Presentan diferentes formas y tamaño; se puede encontrar desde una única lesión hasta múltiples lesiones en forma de manto. Su historia natural no está bien documentada en cuanto a la posibilidad de evolución a displasia o cáncer, no obstante, en la actualidad se reconoce su asociación con mayor riesgo de cáncer (OR 2,4). La vigilancia colonoscópica debe efectuarse a intervalos cortos o en algunos casos puede evaluarse la polipectomia endoscópica o bien la colectomía en el caso de que la vigilancia no sea factible con seguridad.
Estenosis en la colitis ulcerosa:
Una estenosis en la colitis ulcerosa es indicación firme de colectomía, debido al porcentaje elevado de asociación de un carcinoma subyacente.55,56 Puede intentarse trasponer la estenosis con el colonoscopio pediátrico tomando múltiples biopsias e, incluso, realizar un cepillado para citología en el sitio de la estenosis. Aun cuando estas muestras sean negativas para cáncer o displasia, el paciente tiene una probabilidad muy alta de presentar un cáncer, por lo que debe repetirse la colonoscopia en 3 a 4 meses. Puede evaluarse complementariamente realizar una tomografía computada. Siempre esta situación debe poner en consideración el diagnóstico de enfermedad de Crohn en lugar de colitis ulcerosa.
Estenosis en la colitis de Crohn
Si la estenosis es franqueable con el colonoscopio, lo aconsejable es reevaluar la misma mediante una nueva endoscopia al año. Si no es posible, puede intentarse con un equipo pediátrico, de lo contrario se debe evaluar el colon proximal con un colon por enema doble contraste o una tomografía computada. En los pacientes con enfermedad de 20 años de evolución, la posibilidad de un cáncer colorrectal concomitante asciende al 12%, por lo que debe considerarse la cirugía (colectomía total o resección segmentaria) sobre todo si es imposible evaluar el colon proximal a la estenosis.54 Es aconsejable derivar a un centro especializado. La dilatación endoscópica con balón es una posibilidad a considerar sobre todo en casos de estenosis cortas de la enfermedad de Crohn (<6 cm).57 La dilatación de una estenosis larga presenta un mayor riesgo de perforación.
Estenosis anal
En el caso de una estenosis anal, debe realizarse un examen bajo anestesia para descartar malignidad, dado el riesgo elevado de cáncer en esta situación.37 No hay datos bibliográficos para sustentar el intervalo recomendable para el control, por lo que el grupo de expertos que diseñó estas normas consensuó provisoriamente un examen anual bajo anestesia.
Nuevas técnicas endoscópicas para la detección de displasia
- Colonoscopia con luz blanca estándar y luz blanca de alta definición: La gran mayoría de las lesiones displásicas y neoplásicas en EII pueden ser detectadas con colonoscopios de luz blanca estándar, tanto las lesiones planas como elevadas. Esto se basa en los hallazgos de los estudios de Rutter46 y Rubin,44 quienes reportaron una tasa de detección de displasia de 77,3% y 71,8% y de detección de neoplasia de 89,3% y 100% respectivamente. La colonoscopia con luz blanca de alta definición (es decir una imagen con resolución de 850.000 a 1.000.000 de pixeles, versus 100.000 a 400.000 pixeles de los colonoscopios estándar) agrega más sensibilidad para la detección de displasia, sumando una relación de prevalencia ajustada de 2,99.58
- Cromoendoscopia: La cromoendoscopia es la aplicación de tintes tópicos sobre la mucosa colónica para mejorar la delineación y detección de irregularidades de su superficie. Los tintes más usados son el azul de metileno, que es absorbido por los colonocitos normales, no así por las células displásicas o inflamadas por lo que resalta el “pit pattern” de las lesiones mucosas, y el índigo carmín que no es absorbible y decanta hacia las criptas, demarcando las lesiones neoplásicas. Estos tintes se aplican a toda la mucosa colónica, mediante un catéter spray o inyectándolos directamente por el canal de trabajo. No hay estudios que comparen la eficacia entre ambos tintes.58 A la fecha existe bastante evidencia que avala la efectividad del uso de cromoendoscopia en EII. Un estudio realizado por Kiesslich y col.59 comparó colonoscopía convencional versus cromoendoscopia usando azul de metileno en 165 pacientes con CU de larga data, estimando un aumento de tres veces en la detección de neoplasia intraepitelial (p=0,003), permitiendo diferenciar entre lesiones neoplásicas y no neoplásicas con una sensibilidad y especificidad del 93%. Rutter y col.46 en un estudio de 100 pacientes, logró diagnosticar siete casos de displasia adicional con cromoendoscopia.Un meta-análisis reciente que incluyó seis estudios con un total de 1.277 pacientes, estimó la diferencia global de detección de displasia entre colonoscopía regular y cromoendoscopia de 7%. Es importante destacar que el aumento del diagnóstico de lesiones displásicas planas fue de 27%.60 Un segundo meta-análisis estimó la correlación de detección por cromoendoscopia de neoplasia intraepitelial-histología en un 83.8% de sensibilidad y un 91.3% de especificidad.61 Las desventajas de la cromoendoscopia son la necesidad de un entrenamiento en aplicación de tinciones, análisis de “pit pattern” e identificación de lesiones, especialmente planas, y un mayor tiempo de examen, estimándose en promedio 11 minutos adicionales, que podrían compensarse eliminando el protocolo de toma de biopsias escalonadas.58 Respecto a las recomendaciones de guías internacionales para cromoendoscopia, ya mencionamos que el grupo ECCO la prefiere sobre las biopsias escalonadas. Las guías AGA del año 2010 reconocen su superioridad en detectar displasias, sin embargo consideran que está sujeta a la experiencia del endoscopista y que por el momento recomiendan mantener el sistema de biopsias por segmentos. Las últimas guías disponibles (ASGE), ya sitúan a la cromoendoscopia como el método de elección para vigilancia sobre cualquier otro.
- FICE y i-Scan: Estas dos técnicas son métodos de cromoendoscopia virtual, sin la necesidad de aplicar tinciones. FICE (Fujinon® Intelligent Chromoendoscopy, Tokyo, Japón) y i-Scan (Pentax®, Tokyo, Japón) funcionan en base a un algoritmo computarizado que procesa la imagen después de ser captada con luz blanca regular. No existen hasta la fecha estudios realizados de detección de displasia/neoplasia en EII con estos métodos.62
- NBI o Narrow Band Imaging: NBI es una tecnología que resalta la estructura de los vasos y la arquitectura de las criptas, usando filtros de luz especializados que modulan la intensidad de los constituyentes del espectro de luz blanca. Existen tres estudios en vigilancia en EII con endoscopios de primera generación y segunda generación (incluyendo de alta resolución), sin encontrar beneficio en la detección de displasia, con una mayor tasa de falsos positivos en biopsias y de lesiones no vistas en la colonoscopía (OR 4.21).50,58 Las últimas guías ASGE no la recomiendan, situando incluso la colonoscopía con luz blanca de definición estándar por sobre la NBI.
- AFI o Colonoscopia con autofluorescencia: La colonoscopía con autofluorescencia usa los espectros de emisiones diferenciales de los tejidos para construir imágenes símiles a la cromoendoscopia. La presencia de fluoróforos en los colonocitos permite que al aplicar una luz de corta longitud, la mucosa emita (autofluorescencia) una mayor longitud de onda. La diferencia entre ambas entrega información acerca de si esa mucosa es normal, presenta inflamación, hiperplasia o displasia. Si bien ha mostrado resultados prometedores en la detección de displasia en EII, hasta el momento se ha restringido a centros altamente especializados y de investigación.58
- CLE o Endomicroscopia láser confocal: Esta técnica permite el examen microscópico de la mucosa in vivo, requiriendo el uso adicional de contraste, siendo la fluoresceína la más utilizada. Por su nivel de magnificación, examina un pequeño porcentaje de la mucosa colónica, por lo que se reserva para el análisis de lesiones sospechosas detectadas por otros métodos, por ejemplo combinada con cromoendoscopia. Sin embargo es altamente operador dependiente, para endoscopistas avanzados y formados en esta técnica, no pudiendo ser recomendada actualmente en forma masiva.58
BIBLIOGRAFÍA
- Itzkowitz, S. H.; Harpaz, N. Diagnosis and management of dysplasia in patients with inflammatory bowel diseases. Gastroenterology 2004;126:1634-1648.
- Eaden, J. A.; Abrams, K.; Mayberry, J. F. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 2001; 48:526-535.
- Ransohoff, D. F. Colon cancer in ulcerative colitis. Gastroenterology 1988; 94:1089-1091.
- Greenstein, A. J.; Sachar, D. B.; Smith, H.; Janowitz, H. D.; Aufses, A. H. Jr. Cancer. Patterns of neoplasia in Crohn’s disease and ulcerative colitis 1980; 46:403-407.
- Ekbon, A.; Helmick, C.; Zack, M.; Adami, H. Ulcerative colitis and colorectal cancer. A population based study. N England J Med 1990; 323:1228.
- Riddell, R. H. Screening strategies in gastrointestinal cancer. Scand J Gastroenterol Suppl 1990; 175:177-184.
- Eaden, J. A.; Mayberry, J. F. British Society for Gastroenterology. Association of Coloproctology for Great Britain and Ireland. Guidelines for screening and surveillance of asymptomatic colorectal cancer in patients with inflammatory bowel disease. Gut 2002; 51:10-12.
- Moum, B.; Ekbom, A.; Vatn, M. H.; Elgjo, K. Change in the extent of colonoscopic and histological involvement in ulcerative colitis over time. Am J Gastroenterol 1999; 94:1564-1569.
- Rhodes, J. M. Ulcerative colitis extent varies with time but endoscopic appearances may be deceptive. Gut 2001; 49:322-323.
- Mathy, C.; Schneider, K.; Chen, Y. Y.; Varma, M.; Terdiman, J. P.; Mahadevan, U. Gross versus microscopic pancolitis and the occurrence of neoplasia in ulcerative colitis. Inflamm Bowel Dis 2003; 9:351-355.
- Brentnall, T. A.; Haggitt, R. C.; Rabinovitch, P. S. et al. Risk and natural history of colonic neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis. Gastroenterology 1996; 110:331-338.
- Broome, U.; Lofberg, R.; Veress, B. et al. Primary sclerosing cholangitis and ulcerative colitis. Evidence for increased neoplastic potential. Hepatology 1995; 22:1404-1408.
- Terg R, Sambuelli A, Coronel y colaboradores. Prevalence of primary sclerosing cholangitis in patients with ulcerative colitis and the risk of developing malignancies. A large prospective study. Acta Gastroenterol Latinoam 2008;38:26-33.
- Loftus, E. V. Jr; Sandborn, W. J.; Tremaine, W. J.; Mahoney, D. W.; Zinsmeister, A. R.; Offord, K. P.; Melton, L. J. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis. Gastroenterology 1996; 110:432-440.
- Loftus, E. V. Jr; Aguilar, H. I.; Sandborn, W. J.; Tremaine, W. J.; Krom, R. A.; Zinsmeister, A. R.; Graziadei, I. W.; Wiesner, R. H. Risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis following orthotopic liver transplantation. Hepatology 1998; 27:685-690.
- Nuako, K. W.; Ahlquist, D. A.; Mahoney, D. W.; Schaid, D. J.; Siems, D. M.; Lindor, N. M. Familial predisposition for colorectal cancer in chronic ulcerative colitis: a case-control study. Gastroenterology 1998; 115:1079-1083.
- Askling, J.; Dickman, P. W.; Karlen, P. et al. A. Family history as a risk factor for colorectal cáncer in inflammatory bowel disease. Gastroenterology 2001; 120:1356-1362.
- Eaden, J. A.; Mayberry, J. F. Colorectal cancer complicating ulcerative colitis: a review. Am J Gastroenterol 2000; 95: 2710-2719.
- Prior, P.; Gyde, S. N.; Macartney, J. C.; Thompson, H.; Waterhouse, J. A.; Allan, R. N. Cancer morbidity in ulcerative colitis. Gut 1982; 23:490-497.
- Rutter, M.; Saunders, B.; Wilkinson, K.; Rumbles, S.; Schofield, G.; Kamm, M.; Williams, C.; Price, A.; Talbot, I.; Forbes, A. Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 2004; 126:451-459.
- Eaden, J.; Abrams, K.; Ekbom, A.; Jackson, E.; Mayberry, J. Colorectal cancer prevention in ulcerative colitis: a case-control study. Aliment Pharmacol Ther 2000; 14:145-153.
- Velayos FS, Terdiman JP, Walsh JM. Effect of 5-aminosalicylate use on colorectal cancer and dysplasia risk: a systematic review and metaanalysis of observational studies. Am J Gastroenterol. 2005;100:1345-53.
- Carrat, F.; Seksik, P.; Bouvier, A. M. et al. Aminosalicylates, Thiopurines and the Risk of Colorectal Cancer in Inflammatory Bowel Diseases: A Case-Control Study Nested in the CESAME Cohort. Gastroenterology 2010; 138 S 47.
- Connell, W. R.; Kamm, M. A.; Dickson, M.; Balkwill, A. M.; Ritchie, J. K.; Lennard-Jones, J. E. Long-term neoplasia risk after azathioprine treatment in inflammatory bowel disease. Lancet 1994; 343:1249-1252.
- Jess T, Lopez A, Andersson M, Beaugerie L, Peyrin-Biroulet L: Thiopurines and risk of colorectal neoplasia in patients with inflammatory bowel disease: a meta-analysis. Clin Gastroenterol Hepatol. 2014 Nov;12(11):1793-1800.
- Actis GC, Pellicano R, David E, Sapino A.Azathioprine, mucosal healing in ulcerative colitis, and the chemoprevention of colitic cancer: a clinical-practice-based forecast. Inflamm Allergy Drug Targets. 2010 Mar;9(1):6-9.
- Lashner, B. A.; Provencher, K. S.; Seidner, D. L.; Knesebeck, A.; Brzezinski, A. The effect of folic acid supplementation on the risk for cancer or dysplasia in ulcerative colitis. Gastroenterology 1997; 112:29-32.
- Bernstein, C. N.; Blanchard, J. F.; Metge, C.; Yogendran, M. Does the use of 5-aminosalicylates in inflammatory bowel disease prevent the development of colorectal cancer? Am J Gastroenterol 2003; 98:2784-2788.
- Tung, B. Y.; Emond, M. J.; Haggitt, R. C.; Bronner, M. P.; Kimmey, M. B.; Kowdley, K. V.; Brentnall, T. A. Ursodiol use is associated with lowerprevalence of colonic neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med 2001; 134:89-95.
- Pardi, D. S.; Loftus, E. V. Jr; Kremers, W. K.; Keach, J.; Lindor, K. D. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology 2003; 124:889-893.
- Morson, B. C.; Pang, L. S. C. Rectal biopsy is an aid to cancer control in ulcerative colitis. Gut 1967; 8:423-424.
- Riddell, R. H.; Goldman, H.; Ransohoff, D. F.; Appelman, H. D.; Fenoglio, C. M.; Haggit, R. C.; Ahren, C.; Correa, P.; Hamilton, S. R.; Morson, B. C.; Sommers, S. C.; Yardley, J. H. Dysplasia in inflammatory bowel disease: Standardized clasiffication with provisional clinical implications.Hum Pathol 1983; 14: 931-938.
- Lennard-Jones, J. E.; Melville, D. M.; Morson, B. C.; Ritchie, J. K.; Williams, C. B. Precancer and cancer in extensive ulcerative colitis: findings among 401 patients over 22 years. Gut 1990; 31:800-806.
- Bernstein, C. N.; Shanahan, F.; Weinstein, W. M. Are we telling patients the truth about surveillance colonoscopy in ulcerative colitis? Lancet 1994; 343:71-74.
- Dixon, M. F.; Brown, L. J.; Gilmour, H. M.; Price, A. B.; Smeeton, N. C.; Talbot, I. C.; Williams, G. T. Observer variation in the assessment of dysplasia in ulcerative colitis. Histopathology 1988; 13:385-397.
- Melville, D. M.; Jass, J. R.; Morson, B. C.; Pollock, D. J.; Richman, P. I.; Shepherd, N. A.; Ritchie, J. K.; Love, S. B.; Lennard-Jones, J. E. Observer study of the grading of dysplasia in ulcerative colitis: comparison with clinical outcome. Hum Pathol 1989; 20:1008-1014.
- Connell, W. R.; Lennard-Jones, J. E.; Williams, C. B.; Talbot, I. C.; Price, A. B.; Wilkinson, K. H. Factors affecting the outcome of endoscopic surveillance for cancer in ulcerative colitis.Gastroenterology 1994; 107:934-944.
- Ullman, T. A.; Croog, T.; Harpaz, N.; Sachar, D.; Itzkowitz, S. Progression of flat low-grade dysplasia to advanced neoplasia in patients with ulcerative colitis. Gastroenterology 2003; 125:1311-1319.
- Befrits, R.; Ljung, T.; Jaramillo, E.; Rubio, C. Low-grade dysplasia in extensive, long-standing inflammatory bowel disease: a follow-up study. Dis Colon Rectum 2002; 45:615-620.
- Lim, C. H.; Dixon, M. F.; Vail, A.; Forman, D.; Lynch, D. A.; Axon, A. T. Dysplasia in ulcerative colitis. Histopathology 1988; 13:385-397. Ten year follow up of ulcerative colitis patients with and without low grade dysplasia. Gut 2003; 52:1127- 1132.
- Rubin, P. H.; Friedman, S.; Harpaz, N.; Goldstein, E.; Weiser, J.; Schiller, J.; Waye, J. D.; Present, D. H. Colonoscopic polypectomy in chronic colitis: conservative management after endoscopic resection of dysplastic polyps. Gastroenterology 1999; 117:1295-1300.
- Odze, R. D.; Farraye, F. A.; Hecht, J. L.; Hornick, J. L. et al. Longterm follow-up after polypectomy treatment for adenomalike dysplastic lesions in ulcerative colitis. Clin Gastroenterol Hepatol 2004; 2:534-541.
- Cairns SR, Scholefield JH, Steele RJ, Dunlop MG, Thomas HJ, Evans GD, Eaden JA, Rutter MD, Atkin WP, Saunders BP, et al. Guidelines for colorectal cancer screening and surveillance in moderate and high risk groups (update from 2002) Gut. 2010;59:666–689.
- Rubin, C. E.; Haggitt, R. C.; Burmer, G. C.; Brentnall, T. A.; Stevens, A. C.; Levine, D. S.; Dean, P. J.; Kimmey, M.; Perera, D. R.; Rabinovitch, P. S. DNA aneuploidy in colonic biopsies predicts future development of dysplasia in ulcerative colitis. Gastroenterology 1992; 103:1611-1620.
- Van den Broek FJ, Stokkers PC, Reitsma JB, Boltjes RP, Ponsioen CY, Fockens P et al. Random biopsies taken during colonoscopic surveillance of patients with longstanding ulcerative colitis: low yield and absence of clinical consequences. Am J Gastroenterol 2014; 109, n.5: 715-22.
- Rutter, M. D.; Saunders, B. P.; Schofield, G. et al. Pancolonic indigo carmine dye spraying for the detection of dysplasia in ulcerative colitis. Gut 2004; 53:256-260.
- Laine L, Kaltenbach T, Barkun A, McQuaid K, Subramanian V, Soetikno R et al. SCENIC international consensus statementon surveillance and management of displasia in inflammatory bowel disease. Gastrointestinal Endoscopy 2015;81:489-501.
- The Paris classification of superficial neoplastic lesions: esophagus, stomach, and colon. Gastrointest Endosc 2003;58(suppl):S3-43.
- Farraye FA, Odze RD, Eaden J, Itzkowitz SH, McCabe RP, Dassopoulos T et al. AGA Institute Medical Position Panel on Diagnosis and Management of Colorectal Neoplasia in Inflammatory Bowel Disease. Gastroenterology 2010 Feb;138,n.2:738-45.
- Annese V, Daperno M, Rutter M, Amiot A, Bossuyt P, East J et al. European evidence based consensus for endoscopy in inflammatory bowel disease. Journal of Crohn’s and Colitis 2013; 7, n.12: 982–1018.
- Woolrich, A. J.; DaSilva, M. D.; Korelitz, B. I. Surveillance in the routine management of ulcerative colitis: the predictive value of low-grade dysplasia. Gastroenterology 1992; 103: 431-438.
- Winawer, S.; Fletcher, R.; Rex, D.; Bond, J. R.; Ferrucci, J.; Ganiats, T.; Levin, T.; Woolf, S.; Johnson, D.; Kirk, L.; Litin, S.; Simmang, C.; Gastrointestinal Consortium Panel et al. Colorectal cancer screening and surveillance: clinical guidelines and rationale-update based on new evidence. Gastroenterology 2003; 124:544-560.
- Sachar, D. B. Cancer in Crohn’s disease: dispelling the myths. Gut 1994; 35:1507-1508.
- Friedman, S.; Rubin, P. H.; Bodian, C. et al. Screening and surveillance colonoscopy in chronic Crohn’s colitis. Gastroenterology 2001; 120:820-826
- Gumaste, V.; Sachar, D. B.; Greenstein, A. J. Benign and malignant colorectal strictures in ulcerative colitis. Gut 1992; 33:938-941.
- Reiser, J. R.; Waye, J. D.; Janowitz, H. D. et al. Adenocarcinoma in strictures of ulcerative colitis without antecedent dysplasia by colonoscopy. Am J Gastroenterol 1994; 89:119-122.
- Saunders, B. P.; Brown, G. J.; Lemann, M. et al. Balloon dilation of ileocolonic strictures in Crohn’s disease. Endoscopy 2004; 36:1001-1007.
- Naymagon S, Marion J. Surveillance in Inflammatory Bowel Disease, Chromoendoscopy and Digital Mucosal Enhancement. Gastrointest Endoscopy Clin N Am 2013; 23: 679–694
- Kiesslich, R.; Fritsch, J.; Holtmann, M. et al. Methylene blueaided chromoendoscopy for the detection of intraepithelial neoplasia and colon cancer in ulcerative colitis. Gastroenterology 2003; 124:880-888.
- Subramanian V, Mannath J, Ragunath K, Hawkey CJ. Metaanalysis: the diagnostic yield of chromoendoscopy for detecting dysplasia in patients with colonic inflammatory bowel disease. Aliment Pharmacol Ther 2011;33, n.3:304–12.
- Wu L, Li P, Wu J, Cao Y, Gao F. The diagnostic accuracy of chromoendoscopy for dysplasia in ulcerative colitis: metaanalysis of six randomized controlled trials. Colorectal Dis 2012;14:416–20.
- Modha K, Navaneethan U. Advanced therapeutic endoscopist and inflammatory bowel disease: dawn of a new role. World J Gastroenterol 2014; 20, n.13: 3485-3494.