Adenocarcinoma del Intestino Delgado Asociado al Síndrome de Lynch
Adenocarcinoma del Intestino Delgado Asociado al Síndrome de Lynch
Sebastián A. Justo, Rodolfo C. Zannoli, Laura Svidler López, Gabriela L. Sidra, Gastón M. Omeñuk, Julieta Ripamonti, Rita L. O. Pastore
División Cirugía, Hospital Juan A. Fernández, Buenos Aires.
Recibido 30 de enero de 2014
Corregido y aceptado para publicación 19 de marzo de 2014
Correspondencia:
Esta dirección de correo electrónico está siendo protegida contra los robots de spam. Necesita tener JavaScript habilitado para poder verlo.
RESUMEN
El síndrome de Lynch (SL) es una enfermedad autosómica dominante causada por una mutación en los genes de reparación del ADN que predispone al cáncer colorrectal (CCR) y a otros tumores extracolónicos. Entre estos predominan los del endometrio, estómago, tracto urinario alto y ovario. La incidencia de los tumores de intestino delgado (TID) si bien es baja (0,4-2,9%), supera en más de 25 veces a la de la población general. El objetivo de este trabajo es presentar un caso de carcinoma del duodeno en un paciente masculino de 47 años con criterios de Amsterdam II, y discutir las características de los TID en el SL.
Palabras Claves: Tumor del Intestino Delgado; Síndrome de Lynch
ABSTRACT
Lynch Syndrome (LS) is an autosomal dominant condition caused by mutations in the mismatch repair genes that predispose to colorectal cancer (CRC) and other extracolonic tumors. Among these, endometrial, gastric, ovarian, and urinary tract tumors are the commonest. The incidence of small bowel tumors (SBT), although low (0.4-2.9%), exceeds in more than 25 times that of the general population. The purpose of this paper is to communicate a case of carcinoma of the duodenum in a 47 years old male with Amsterdam II criteria, and discuss the characteristics of SBT in LS.
Key words: Small Bowel Tumor; Lynch Syndrome
INTRODUCCIÓN
El síndrome de Lynch (SL) o Cáncer Colorrectal Hereditario No Asociado a Poliposis (CCHNAP) es una enfermedad autosómica dominante causada por una mutación en los genes de reparación del ADN, que predispone al cáncer colorrectal (CCR) y a otros tumores extracolónicos. Se han descripto mutaciones en la línea germinal de diversos genes; MLH1, MSH2, PMS2 y MSH6, siendo las más frecuentes las dos primeras (>85%).
Diversos autores comunicaron su experiencia mostrando que la localización colorrectal es la más frecuente (61,3-67%) y que de las extracolónicas, la predominante es la del endometrio (7-13%); otros órganos afectados son: estómago (3,9%-5,9%), ovario (0,7-3%), tracto urinario alto (1-4,7%), cerebro (1-1,7%), páncreas y tracto hepatobiliar (1-3%) y piel (1,4%).1-3
Un estudio de cohorte de 360 miembros de 50 familias portadoras de mutaciones genéticas del SL, mostró un riesgo relativo de padecer cáncer mucho más alto que el de la población general, con la siguiente distribución: colorrectal [68; intervalo de confianza (IC) 95%, 56 a 81], endometrial (62; IC 95%, 44 a 86), ovárico (13; 95% IC 5,3 a 25), gástrico (6,9; 95% IC 3,6 a 12), tracto biliar (9,1; 95% IC 1,1 a 33), uro-epitelial (7,6; 95% IC 2,5 a 18), riñón (4,7; 95% IC 1 a 14) cerebro (4,5; 95% IC 1,2 a 12). En esta serie no se observó ningún tumor de intestino delgado (TID) en portadores de mutaciones genéticas, mientras que la incidencia acumulativa fue de 82, 60, 13 y 12% para el CCR, endometrial, gástrico y ovárico, respectivamente.4 La incidencia de TID en este síndrome es del 0,4-2,9%,1-3,5 representando un riesgo relativo >25 veces el de la población general.5
A pesar que en el SL los tumores extracolónicos se presentan más comúnmente en los portadores de mutaciones del MSH2, los TID suelen estar más asociados con mutaciones del MLH1, habiendo descripto Vasen y col.6 un riesgo acumulativo del 7% en quienes poseen esta mutación.
El objetivo de este trabajo es comunicar un caso de TID por SL, dada su baja frecuencia.
CASO CLÍNICO
Varón de 47 años con criterios de Amsterdam II para SL tipo 2, por antecedentes familiares (padre fallecido a los 60 años por cáncer del colon sigmoides, hermano de 48 años operado por tumor sincrónico del colon transverso y del recto). Consultó por dolor abdominal, nauseas, vómitos, alteración del ritmo evacuatorio (diarrea/constipación) y pérdida de 35 kg en los últimos 4 meses. Al examen físico se constató desnutrición grave, palidez, y dolor a la palpación en el hemiabdomen superior. Analítica: ligera anemia (Hb 11g/dl, Hto 35%). Seriada gastroduodenal: dilatación del marco duodenal con estenosis de la cuarta porción y de la primera asa yeyunal (Fig.1). Tomografía computada toraco-abdomino-pelviana: distensión gástrica y de la 1a, 2a y 3a porción duodenal, con disminución de la luz de la 4a porción (Fig. 2). Videoendoscopía digestiva alta: progresión hasta la 2a porción duodenal sin lesiones mucosas. Enteroscopía: sin posibilidades de progresar más allá del estómago por presentar gran contenido gástrico. Videocolonoscopía: hasta el ciego sin lesiones mucosas.
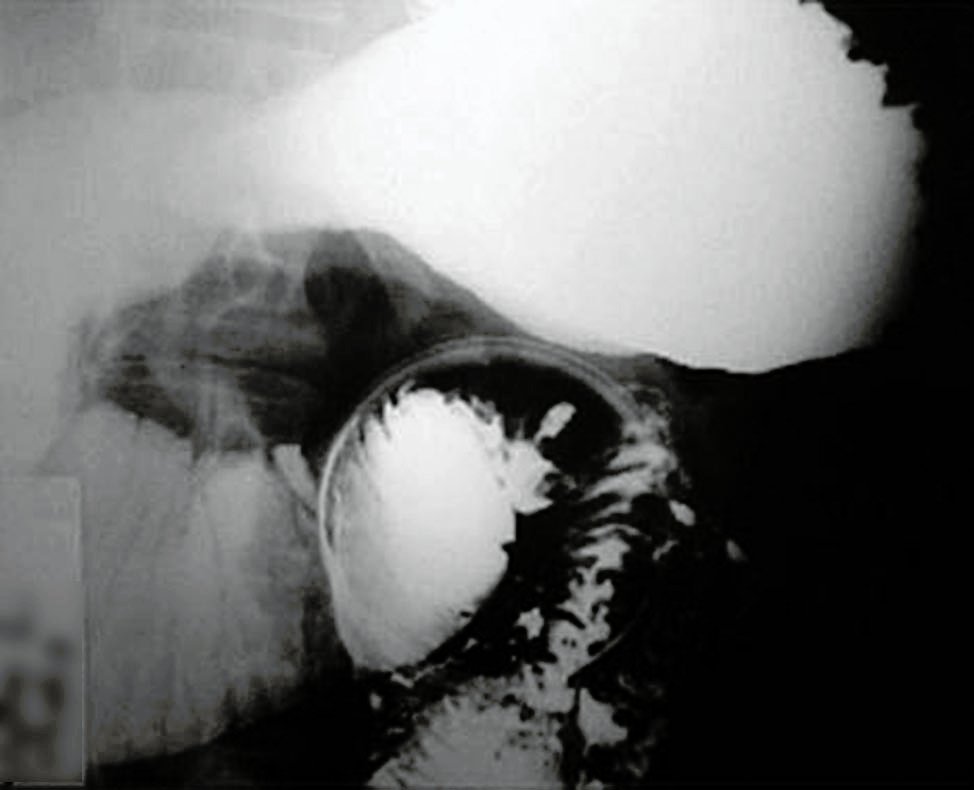
Figura 1: Seriada gastroduodenal que muestra dilatación del marco duodenal con estenosis de la cuarta porción y de la primera asa yeyunal.
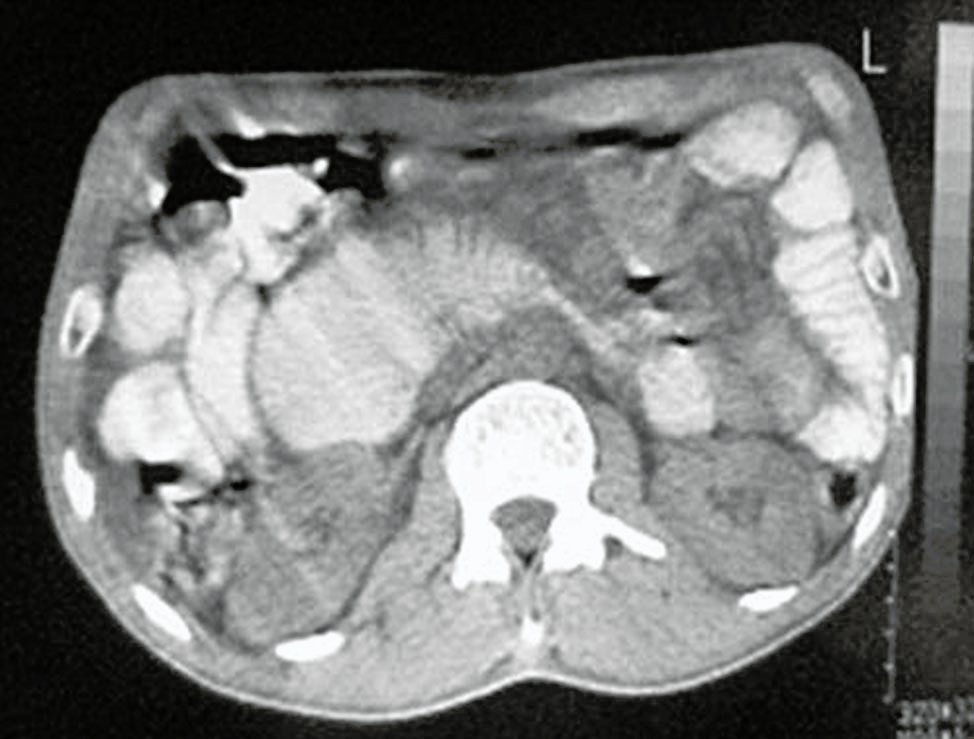
Figura 2: Tomografía computada en la que se observa dilatación duodenal proximal a la estenosis de la cuarta porción.
Con el diagnóstico presuntivo de TID se decide cirugía. Se aborda el abdomen por vía convencional, se identifica compromiso tumoral de la cuarta porción del duodeno y el ángulo de Treitz, con invasión del mesocolon y de la arteria mesentérica superior. Se reseca en bloque el ángulo de Treitz incluyendo la 4a porción duodenal y primera asa yeyunal (Fig. 3), con una anastomosis latero-lateral entre la 3a porción duodenal y el yeyuno. Reimplante de la arteria mesentérica superior en la aorta. Se reseca un segmento del colon transverso y se confecciona una colostomía transversa terminal y una fístula mucosa. Yeyunostomia de alimentación. Anatomía patológica: adenocarcinoma moderadamente diferenciado con infiltración del intestino delgado y el colon, con metástasis ganglionares. Márgenes libres.
Ingresó inicialmente a UTI, donde presentó episodios de diarrea por 14 días atribuida a atrofia vellositaria, tratada con nutrición parenteral. Egresó a los 33 días tolerando dieta oral. Reingresó a los 4 meses por un cuadro de neumonía con derrame, con rescate de pseudomona en el líquido del lavado bronquial, tratada con antibióticos y avenamiento pleural. Evoluciono al shock séptico con óbito a los 35 días.
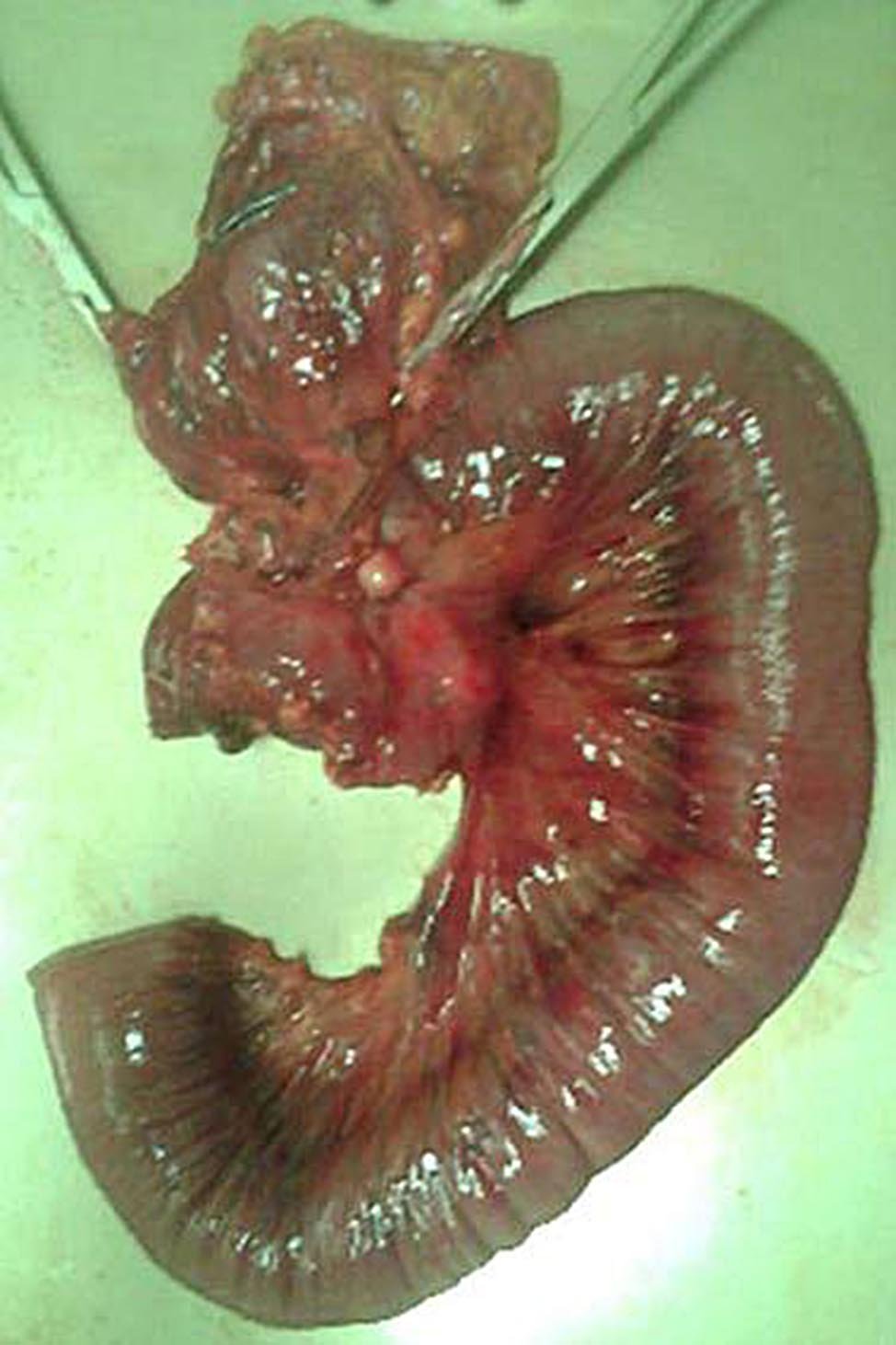
Figura 3: Pieza de resección que incluye en bloque el ángulo de Treitz con la 4a porción duodenal y la primera asa yeyunal, además de parte del colon transverso comprometidos por la lesión tumoral.
DISCUSIÓN
Al igual que los esporádicos, los TID en el SL son poco frecuentes y no difieren en cuanto a la localización, sintomatología, métodos de diagnóstico y tratamiento. Se diferencian en la edad de presentación, las variantes histopatológicas, la frecuencia de tumores sincrónicos y metacrónicos y el pronóstico.
Con respecto a la localización, excepto por la experiencia de Lynch y col.7 que comunicaron la mayor frecuencia en el yeyuno, las otras series publicadas coinciden en señalar al duodeno como el sitio más afectado (36-50% de los casos), seguido del yeyuno y del íleon.8,9
Las formas de presentación clínica son inespecíficas y pueden incluir dolor abdominal, nauseas, vómitos, hemorragia, debilidad, pérdida de peso, anemia e ictericia por obstrucción de la ampolla de Vater.
Estos tumores deben sospecharse en el contexto de antecedentes familiares con criterios clínicos de Amsterdam o Bethesda. Al respecto, se ha descripto que el 50-81% de los pacientes provienen de familias que cumplen los criterios de Amsterdam8,9 y el 47% al menos un criterio de Bethesda.9 El TID fue la primera localización tumoral en 45-57% de los pacientes con estos criterios.8,9
Las mutaciones identificadas en las líneas germinales de los genes reparadores, se presentaron en MLH1 (59-60%), en MSH2 (37-40%) y en MSH6 (4%).8,9
Es interesante destacar que en el 33% de los pacientes el intestino delgado fue el único sitio de malignidad y que 43% de los casos ocurrieron luego de otra localización tumoral casi 14 años después, datos obtenidos de un cuestionario respondido por 11 centros de EEUU, Europa y Australia que forman parte del grupo colaborador internacional sobre CCHNAP.8
La edad de presentación de los TID esporádicos tiene un pico de incidencia en la sexta década de la vida, siendo en el SL más temprana. Lynch y col.7 publicaron en 1989 una serie de 9 casos en 8 familias, en los cuales la edad promedio fue de 47 (31-56) años. Coincidentemente, Rodríguez-Bigas y col.8 en representación del grupo colaborador sobre CCHNAP, comunicaron en 1998 las características de los TID en 42 individuos de 40 familias con CCHNAP que desarrollaron 49 tumores (42 primarios y 7 metacrónicos), cuya edad promedio de presentación fue de 49 años. Sin embargo, Schulmann y col.,9 en un trabajo más reciente (2005), analizaron 32 pacientes del grupo alemán de estudio sobre CCHNAP, donde la edad mediana fue de 39 (15-73) años, con solo un paciente menor de 30 años. Esto es una presentación 10 años menor que la reportada previamente para los TID asociados al SL y 20 años más temprana que para TID esporádicos, como ocurre con los CCR.7-9
Al igual que para el CCR, al momento del diagnóstico del TID también se han descripto otros tumores sincrónicos (17%) y metacrónicos (del propio intestino delgado en el 10% de los casos),8 por lo cual ante su diagnóstico se debe enfatizar la pesquisa, tanto en el pre como en el intraoperatorio, de otros tumores en el intestino delgado y en otras localizaciones habituales en este síndrome.
Igualmente, puede evidenciarse que a diferencia de lo que ocurre con el CCR y otros tumores extracolónicos (endometrio, ovario, estómago), lo habitual para los TID es que se presenten en un solo integrante de la familia, ya que una historia familiar positiva de TID solo se presenta en el 6-17% de los pacientes.8,9
Los tipos histológicos que se presentan en los TID esporádicos son adenocarcinomas (40%), tumores carcinoides (29%), linfomas (17%) y sarcomas (12%). En el SL el tipo más frecuentemente encontrado es el adenocarcinoma (92%) y en mucha menor frecuencia tumores carcinoides y adenomas (4%).7,9
El diagnóstico se puede realizar mediante distintos estudios como la endoscopía digestiva alta, enteroscopía, ecografía, enteroclisis, endocápsula y colangiografía retrograda endoscópica en tumores periampulares. En ocasiones el diagnóstico se realiza en oportunidad de una laparotomía exploratoria por oclusión intestinal.7,10,11
Desafortunadamente en los TID el pronóstico no es tan favorable como el de la localización colónica. Lynch y col.,7 comunican una supervivencia a 5 años del 10-20%, cuando existen metástasis ganglionares y/o a distancia al momento del diagnóstico. Otra serie más reciente con 52% (9/17) de los pacientes con tumores avanzados (EIII-IV) comunica una supervivencia estimada a 5 y 10 años del 44% y 33% respectivamente.8 Este pronóstico es mejor que el observado para los TID esporádicos (estadio: I 55%, IIA 49%, IIB 35%, IIIA 31%, IIIB 18%, IV 5%).11
La quimioterapia y radioterapia no han demostrado mejorar la supervivencia a largo plazo, siendo la cirugía la única opción de tratamiento, ya sea con fines curativos o paliativos.7
Las guías de la National Comprehensive Cáncer Network (NCCN) 2012 indican que no existe evidencia clara que apoye la pesquisa del cáncer del intestino delgado asociado al SL, y sugiere realizar esofagogastroduodenoscopía con duodenoscopía extendida (hasta duodeno distal e incluso yeyuno) con intervalos de 2 o 3 años a partir de los 30-35 años. También se considera la endocápsula con el mismo fin.10
CONCLUSIÓN
Los TID asociados al SL son poco frecuentes, se localizan preferentemente en el duodeno, suelen diagnosticarse cuando son sintomáticos, habitualmente se presentan en un solo miembro de la familia, a una edad que precede en 20 años a la de los esporádicos y son casi exclusivamente adenocarcinomas.
En un tercio de los casos son la única manifestación del SL y suelen ser tumores primarios, aunque también pueden presentarse como tumores metacrónicos muchos años después. En algunos casos son sincrónicos de un cáncer colorrectal u otro extracolónico, por lo que debe investigarse su posible presencia.
El diagnóstico y tratamiento no difiere de los esporádicos, pero el pronóstico es mejor. Sin embargo, es peor que el del CCR en este síndrome.
La pesquisa es dificultosa y no hay evidencias que la avalen claramente.
Ante un paciente, particularmente joven, portador de un TID, se impone indagar antecedentes personales o familiares en busca del SL.
BIBLIOGRAFÍA
- Vasen HFA, Offerhaus GJA, den Hartog Jager FCA, et al. The tumor spectrum in hereditary nonpolyposis colorectal cancer: a study of 24 kindreds in the Netherlands. Int J Cancer 1990; 46:31-4.
- Aarnio M, Mecklin KP, Aaltonen A, Nystrom-Lahti MI, Jarvinen HJ. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int. J. Cancer 1995; 64,430-33.
- Da Silva FC, De Oliveira LP, Monteiro Santos R, et al. Frequency of extracolonic tumors in Brazilian families with Lynch syndrome: analysis of a hereditary colorectal cancer institutional registry. Familial Cancer 2010; 9:563-70.
- Aarnio M, Sankila R, Pukkala E, et al. Cancer Risk in Mutation Carriers of DNA-Mismatch-Repair Genes. Int. J. Cancer 1999; 81, 214–18.
- Watson P, Lynch HT. Extracolonic Cancer in Hereditary Nonpolyposis Colorectal Cancer. Cancer 1993; 71:677-685.
- Vasen HFA, Stormorken A, Menko FM, et al. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol 2001; 19:4074-80.
- Lynch HT, Smyrk TC, Lynch PM, et al. Adenocarcinoma of the Small Bowel in Lynch Syndrome. Cancer 1989; 64:2178-83.
- Rodríguez-Bigas MA, Vasen HS, Lynch HT, et al. Characteristics of Small Bowel Carcinoma in Hereditary Nonpolyposis Colorectal Carcinoma. Cancer 1998; 83: 240-44.
- Schulmann K, Brasch FE, Kunstmann E, et al. HNPCC-associated small bowel cancer: clinical and molecular characteristics. Gastroenterology 2005; 128:590–9.
- National Comprehensive Cancer Network. Colorectal Cancer Screening. http://www.trikobe.org/nccn/guideline/colorectal/english/colorectalscreening.pdf
-
American Cancer Society. Small Intestine Cancer. http://www.cancer.org/acs/groups/cid/documents/webcontent/003140-pdf.pdf.